Síguenos en Nuestras Redes Sociales
Jerry R. Mendell · Francesco Muntoni · Craig M. McDonald · Eugenio M. Mercuri · Emma Ciafaloni · Hirofumi Komaki · Carmen Leon‐Astudillo · Andrés Nascimento · Crystal Proud · Ulrike Schara‐Schmidt · Aravindhan Veerapandiyan · Craig M. Zaidman · Matthew Furgerson · Kai Ding · Preeti Singh · Rachael Potter · Damon R. Asher · Alexander P. Murphy · Carol Reid · Gregory Hooper · Carmen O. Torre · Marianna Manfrini · Louise R. Rodino‐Klapac
Recibido: 10 de septiembre de 2025 / Aceptado: 12 de diciembre de 2025
© Los Autores 2026
RESUMEN
Introducción: Delandistrogene moxeparvovec es una terapia génica basada en un vector recombinante del virus adeno-asociado Rh serotipo 74 (rh74) que aborda la ausencia de distrofina funcional en la distrofia muscular de Duchenne (DMD). El ensayo EMBARK es un ensayo de Fase 3, de dos partes, diseño cruzado, aleatorizado, controlado con placebo, que evalúa la seguridad y eficacia de delandistrogene moxeparvovec (una sola dosis intravenosa de 1.33×1014 genomas vectoriales /kg.) en pacientes masculinos ambulatorios con DMD de 4 a <8 años de edad; N=125. Los resultados de un año demostraron la seguridad manejable de delandistrogene moceparvovec, consistente con ensayos clínicos previos. El parámetro principal (el cambio del punto de inicio en los puntajes totales de la escala ambulatoria North Star [NSAA, por sus siglas en inglés] a las 52 semanas en comparación con el placebo) no tuvo importancia estadística. Sin embargo, los parámetros clave secundarios, compuestos de pruebas funcionales cronometradas, sugirieron retraso o estabilización de la progresión de la enfermedad con delandistrogene moxeparvovec, lo cual podría hacerse evidente cada vez más por periodos de tiempo más largos. Reportamos resultados funcionales y de seguridad de 2 años de seguimiento en pacientes que recibieron delandistrogene moxeparvovec en la parte 1 de EMBARK.
Métodos: Como resultado de un estudio de diseño cruzado, los resultados funcionales de 2 años de pacientes que recibieron delandistrogene moxeparvovec en la parte 1 de EMBARK fueron comparados, por análisis pre-específico, con un control externo (CE) ponderado por puntaje de propensión aplicado tras el emparejamiento.
Resultados: A los 2 años, los pacientes del estudio EMBARK mostraron beneficios significativos estadísticamente frente a la cohorte CE en los pronósticos de los resultados funcionales para el retraso de la pérdida de la ambulación (NSAA, el tiempo para levantarse, prueba para caminar de 10 metros), demostrando estabilización mantenida o retraso de la progresión de la enfermedad. La expresión de microdistrofina y la localización sarcolemal con delandistrogene moxeparvovec se mantuvieron por 64 semanas. No se observaron nuevos signos referentes a la seguridad entre la semana 52 y la semana 104. Entre el punto de partida y la semana 104, no hubo fallecimientos relacionados al tratamiento, suspensiones del estudio debido a eventos adversos, o eventos adversos clínicamente significativos mediados por la activación del complemento.
Conclusiones: A los 2 años, se observó la estabilización o retraso de la progresión de la enfermedad en la DMD en pacientes masculinos ambulatorios con DMD de 4 a <8 años de edad que recibieron delandistrogene moxeparvovec frente a la cohorte CE correspondiente. La seguridad fue consistente con los datos de 1 año del estudio EMBARK y manejable con un monitoreo apropiado.
ClinicalTrials.gov: NCT05096221.
Palabras clave: Delandistrogene moxeparvovec; DMD; distrofia muscular de Duchenne; Terapia génica; Enfermedades neuromusculares
| Aspectos clave resumidos |
| ¿Por qué realizar este estudio? |
| El estudio de Fase 3 EMBARK en pacientes masculinos ambulatorios con DMD de 4 a <8 años de edad evaluó delndistrogene moxeparvovec (una terapia génica basada en un vector recombinante del virus adeno-asociado Rh serotipo 74, aprobada en EE. UU. para el tratamiento de pacientes ambulatorios con DMD de 4 años de edad y mayores; también está aprobada en otros países exclusivos). Los resultados de un año del estudio EMBARK para delandistrogene moxeparvovec frente al placebo ya se han publicado previamente. |
| Reportamos aquí los resultados de 2 años provenientes de pacientes tratados con delandistrogene moxeparvovec en el estudio EMBARK frente a una cohorte de control externo (CE) ponderado por puntaje de propensión aplicado tras el emparejamiento (debido al diseño cruzado del estudio y la ausencia de un placebo después de 1 año). |
| ¿Qué se ha sabido del estudio? |
| A los 2 años de seguimiento en el estudio EMBARK, se observó estabilización mantenida o retraso de la progresión de la enfermedad DMD en los pacientes recibiendo delandistrogene mozeparvovec frente al CE correspondiente. La expresión de la microdistrofina fue evidente por 64 semanas. El perfil de seguridad de 2 años fue consistente con los datos de 1 año del estudio EMBARK. |
| Estos hallazgos complementan los datos a largo plazo de delandistrogene moxeparvovec y respaldan el uso continuo de delandistrogene moxeparvovec en el tratamiento de pacientes ambulatorios con DMD. |
____________________________________________________________________________
Presentación previa: presentado previamente en una parte, en el Simposio sobre Intervención Temprana en Duchenne de Parent Project Muscular Dystrophy (PPMD), el 5 de marzo de 2025; la Conferencia Clínica-Científica de la asociación Muscular Dystrophy Association (MDA), en Dallas, Texas, EE. UU, del 16 al 19 de marzo de 2025; la Reunión Anual de la Sociedad Estadounidense de Terapia Génica y Celular (ASGCT, por sus siglas en inglés), en Nueva Orleans, Louisiana, EE. UU., del 13 al 17 de mayo de 2025; el Congreso de la Sociedad Europea de Neurología Pediátrica, en Munich, Alemania, del 8 al 12 de julio de 2025; y el 30o Congreso Internacional Anual de World Muscle Society (WMS), en Viena, Austria, del 7 al 11 de octubre de 2025.
____________________________________________________________________________
Información suplementaria
La versión digital en línea contiene material suplementario disponible en el siguiente enlace:
https://doi.org/10.1007/s40120-025-00879-8
____________________________________________________________________________
J. R. Mendell ()
Centro para Terapia Génica, del Hospital Infantil Nacional, Columbus, Ohio, EE. UU. y Sarepta Therapeutics, Inc, Cambridge, Massachusetts, EE. UU.
e-mail: JMendell@sarepta.com
M. Furgerson · K. Ding · P. Singh · R. Potter · D. R. Asher · L. R. Rodino‐Klapac
Sarepta Therapeutics, Inc, Cambridge, Massachusetts, EE. UU.
F. Muntoni
Centro Neuromuscular Dubowitz, Centro de Investigación Biomédica NIHR Great Ormond Street, Instituto de Salud Infantil e Instituto de Neurología Great Ormond Street, University College London, y Fundación del Hospital Great Ormond Street, London, Reino Unido
C. M. McDonald
UC Davis Health, Sacramento, California, EE. UU.
E. M. Mercuri
Instituto de Neurología Pediátrica, Universidad Pediátrica y Nemo Pediétrico, Fundación Policlinico Gemelli IRCCS, Roma, Italia
E. Ciafaloni
Centro Médico de la Universidad de Rochester, Rochester, Nueva York, EE. UU.
H. Komaki
Centro Médico Translacional, Centro Nacional de Neurología y Psiquiatría, Kodaira, Tokyo, Japón
C. Leon‐Astudillo
Departamento de Pediatría, Universidad de Florida, Gainesville, Florida, EE. UU.
A. Nascimento
Unidad Neuromuscular, Departamento de Neuropediatría, Hospital Sant Joan de Déu, Fundación Sant Joan de Déu, CIBERER – ISC III, Barcelona, España
C. Proud
Hospital Infantil King’s Daughters, Norfolk, Virginia, EE. UU.
U. Schara‐Schmidt
Department of Neurología Pediátrica, Center para Enfermedades Neuromusculares en Niños y Adolescentes, Universidad Clinic Essen, Universidad de Duisburg-Essen, Essen, Alemania
A. Veerapandiyan
Departamento de Pediatría, División of Neurología, Universidad de Arkansas para Ciencias Médicas, Hospital Infantil de Arkansas, Little Rock, Arkansas, EE. UU.
C. M. Zaidman
Departmento de Neurología, Universidad Washington en St Louis, St Louis, Missouri, EE. UU.
A. P. Murphy · C. Reid · G. Hooper · C. O. Torre
Roche Products Ltd, Welwyn Garden City, Reino Unido
M. Manfrini
F. Hoffmann-La Roche Ltd, Basel, Suiza
____________________________________________________________________________
INTRODUCCIÓN
La distrofia muscular de Duchenne (DMD) es una enfermedad neuromuscular asociada al cromosoma X causada por variantes patogénicas en el gen DMD conduciendo a la ausencia de distrofina funcional, una proteína subsarcolemmal clave para la integridad, función, y protección muscular [1,2]. Las personas con DMD experimentan daño progresivo en la función motora, pérdida de la ambulación, y muerte eventual por complicaciones cardíacas y/respiratorias.
Delandistrogene moxeparvovec, una terapia génica basada en un vector de virus adeno-asociado (VAA) (recombinante de VAA Rh serotipo 74), ha sido aprobada en Estados Unidos de América y otros países selectos [3]. Los estudios clínicos en fases iniciales (Estudio 101, NCT03375164, un ensayo de fase 1/2a, abierto, no aleatorio, N=4 [4]; Estudio 102, NCT03769116, un estudio cruzado de dos partes, de Fase 2, aleatorizado, a doble ciego, controlado con placebo, N=41 [5]; cohorte 1 del estudio ENDEAVOR [Estudio 103], NCT04626674, de un solo brazo, abierto, N=20 [6] evaluaron la seguridad, expresión de microdistrofina, y los resultados funcionales en pacientes ambulatorios de ≥4 a <8 años de edad. Los hallazgos generales de estos estudios mostraron que delandistrogene moxeparvovec tiene un perfil beneficio-riesgo favorable basado en un perfil de seguridad manejable, expresión robusta de microdistrofina hasta 60 semanas después del tratamiento, y sugirieron estabilización de resultados funcionales por 4 años. Basado en estos hallazgos, un ensayo extenso cruzado de dos partes, de Fase 3, aleatorizado, controlado con placebo (EMBARK, NCT05096221) se llevó a cabo para evaluar la seguridad y eficacia de delandistrogene moxeparvovec en pacientes ambulatorios de ≥4 a <8 años de edad. Al año de seguimiento de los pacientes tratados en la parte 1 del estudio, delandistrogene moxeparvovec mostró un perfil de seguridad manejable que fue consistente con las fases tempranas de ensayos clínicos. La evaluación funcional al año mostró una diferencia numéricamente favorable pero no estadísticamente significativa en el cambio en la escala ambulatoria North Star (NSAA, por sus siglas en inglés) del punto de inicio (parámetro principal) en comparación con el placebo. Los parámetros secundarios incluyendo el tiempo para levantarse (TTR, por sus iniciales en inglés) y la prueba para caminar de 10 metros (10MWR, por su abreviatura en inglés) y otros parámetros funcionales mostraron efectos favorables del tratamiento en la población general, basados en las diferencias e intervalos de confianza (CI, por sus siglas en inglés) según el método de mínimos cuadrados (LSM, por sus iniciales en inglés) entre grupos, aunque no se puede extraer importancia estadística [7].
Los criterios de inclusión del estudio EMBARK seleccionaron a los pacientes que estaban en la fase de adquisición de habilidades según la NSAA o de estabilidad de la enfermedad de la DMD, con los criterios de admisión del puntaje total de la NSAA siendo entre >16 y <29 puntos, y el TTR de <5 seg. [7]. Se postuló que la medida relativamente aproximada de la NSAA podría no haber sido lo suficientemente sensible en la población de este estudio como para detectar un efecto estabilizador después de 1 año de seguimiento. Sin embargo, los parámetros clave secundarios comprometieron las pruebas de función cronometradas que son potencialmente más sensibles a cambios funcionales en esta población de pacientes, conforme se da la transición gradualmente en la fase de declive de la enfermedad según NSAA. Estos parámetros clave secundarios sugirieron un retraso o estabilización de la progresión de la enfermedad en pacientes recibiendo delandistrogene moxeparvovec. La estabilización de la trayectoria de la enfermedad podría conducir a un aumento de la divergencia del curso esperado del declive progresivo con el tiempo, lo cual podría eventualmente reflejarse en los puntajes totales de la NSAA.
En el brazo (grupo) placebo del estudio EMBARK, los pacientes recibieron placebo por 1 año antes de cruzarse en el grupo con tratamiento y recibir delandistrogene moxeparvovec [7]. Como consecuencia, con la falta de un grupo de control con placebo, la evaluación de la eficacia funcional a largo plazo y de la durabilidad del tratamiento necesita el uso de controles externos (CE) como un comparador del grupo tratamiento con delandistrogene moxeparvocex. Los CE están bien establecidos como herramientas útiles para evaluar los beneficios del tratamiento a largo plazo [8–10]. En el caso de la DMD, como resultado de la heterogeneidad de la enfermedad, es importante utilizar metodologías que crean una cohorte de CE compuesta de pacientes que coinciden con las características pronósticas del punto de inicio y que reflejen el estándar actual del cuidado. Por fortuna, hay una base de datos amplia de CE derivada de estudios de historia natural, ensayos aleatorios, y datos del mundo real que pueden ser utilizados.
Aquí reportamos los resultados del seguimiento de 2 años de un análisis pre-especificado de la eficacia en pacientes tratados con delandistrogene moxeparvovec en la parte 1 del estudio EMBARK comparado con una cohorte CE ponderada por puntaje de propensión aplicado tras el emparejamiento. La cohorte CE consistió de pacientes recibiendo corticosteroides estándar del estudio clínico FOR-DMD (NCT01603407), y los estudios observacionales prospectivos PRO-DMD-01 (NCT01753804) y CINRG DNHS (NCT00468832). Los resultados de la seguridad entre las semanas 52 y 104 para los pacientes tratados con delandistrogene moxeparvovec en la parte 1 también se reportaron.
MÉTODOS
Aprobación ética
El diseño del estudio EMBARK, incluyendo la selección de pacientes, declaraciones de ética (incluyendo una lista completa de comités revisores [ver sección de Material Suplementario] y comités de ética), y descripciones de la terapia génica, ya se ha publicado previamente por completo [7]. Este ensayo fue realizado en cumplimiento de los lineamientos de Buenas Prácticas Clínicas y la Declaración de Helsinki. El comité revisor institucional y el comité de ética en cada sede aprobó el protocolo del ensayo y todas las modificaciones. Los detalles de todos los comités revisores institucionales y comités de ética están disponibles en la sección del Material Suplementario. Según se haga una solicitud razonable, el protocolo del ensayo está disponible por parte del autor correspondiente. El análisis principal del estudio se llevó a cabo después de que todos los pacientes completaran la parte 1, y estos hallazgos han sido publicados [7]. Todos los autores se involucraron en el diseño del estudio, recolección de datos, análisis, e interpretación, así como en la redacción, revisión, y aprobación del manuscrito, y en conjunto estuvieron de acuerdo en su publicación [7]. El patrocinador fue responsable de confirmar el diseño del ensayo final, protocolo, y realización, junto con el mantenimiento de la base de datos, análisis de datos, y precisión de los mismos [7]. Todos los autores recopilaron y tuvieron acceso completo a los datos, y atestiguaron su precisión y completamiento de acuerdo con el protocolo del ensayo [7]. Los padres de familia/tutores legales proporcionaron su consentimiento informado y, cuando se requería, se obtuvo la autorización de los pacientes. La seguridad, eficacia, calidad de los datos, e integridad del estudio continúan siendo monitoreados por un comité monitor de datos independiente [7].
Participantes
Brevemente, los pacientes del estudio EMBARK tenían entre 4 y < 8 años al momento de la aleatorización, presentaban una variante patógena en el gen DMD contenida totalmente entre los exones 18 y 79 (incluídos), tuvieron un puntaje total en la NSAA de >16 y <29 puntos, y un TTR de <5 al momento de la selección, y recibían una dosis diaria estable de corticosteroides orales por al menos 12 semanas antes de la selección, esperando que la dosis y el régimen se mantuvieran constantes (a excepción de las modificaciones para los cambios en el peso) a lo largo del estudio [7]. El estudio EMBARK se realizó entre el 27 de octubre de 2021 y el 25 de octubre de 2024.
Diseño del estudio
Los pacientes fueron aleatorizados (en una relación 1:1) para recibir ya fuera una sola administración intravenosa de delandistrogene moxeparvovec o placebo. En las partes 1 y 2, el día antes de la infusión, todos los pacientes empezaron a recibir corticosteroides (1 mg./kg. al día por al menos 60 días posteriores a la infusión) además de los corticosteroides estándar orales de manera estable que estaban recibiendo [7]. El estudio cruzado consistió de dos partes de 52 semanas (parte 1 y parte 2, ambas completadas) seguidas de un estudio de seguimiento abierto de al menos 5 años [7]. En la parte 2, los pacientes tratados con delandistrogene moxeparvovec recibieron placebo en la parte 1, mientras que los pacientes que recibieron placebo en la parte 1 recibieron delandistrogene moxeparvovec [7].
Se recopilaron biopsias musculares, como se describió anteriormente, de los pacientes en un subgrupo preseleccionado de las sedes (basados en la experiencia al realizar biopsias musculares en su repertorio de diagnóstico de rutina) a las 12 y 64 semanas [7]. La metodología para procesar y analizar las muestras biológicas ya se han publicado [7]. El reporte de los eventos adversos (EA) fue continuo, comenzando con el consentimiento/autorización informada [7].
Cohorte de control externo (CE)
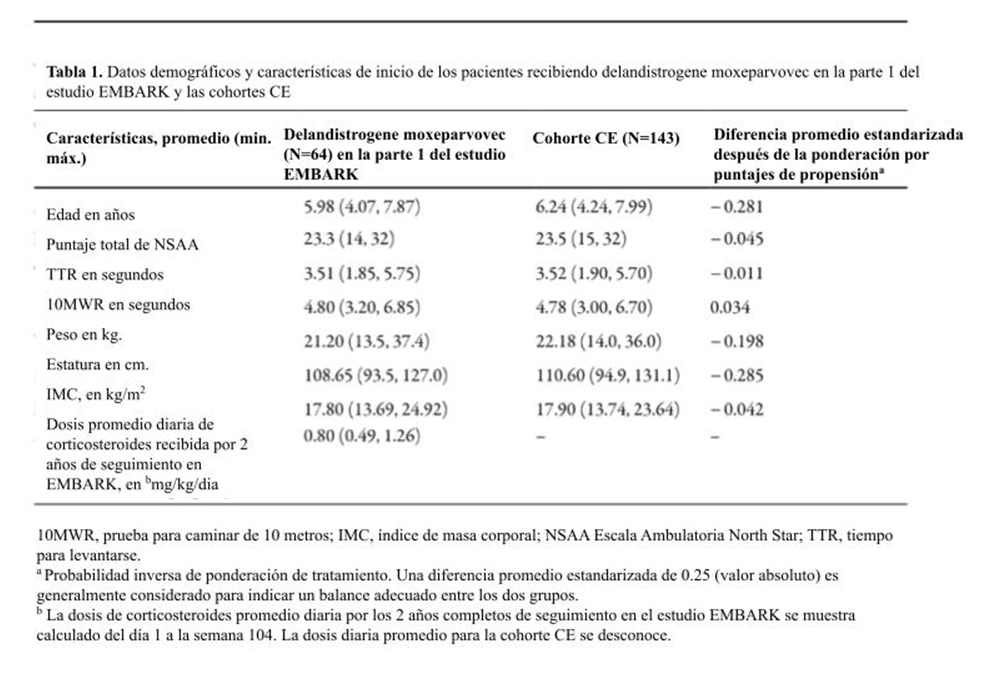
Ante la ausencia de un brazo (grupo) placebo debido al diseño cruzado, en un análisis pre-específico, se compararon los resultados funcionales de 2 años con una cohorte CE de pacientes con DMD usando una ponderación por puntaje de propensión aplicado tras el emparejamiento (ponderación inversa de la probabilidad de tratamiento) [11]. La ponderación por puntaje de propensión se basó en la edad, el puntaje total de la NSAA de inicio, el TTR, 10MWR, estatura, peso, e índice de masa corporal, para asegurar que las características de inicio de los pacientes en la cohorte CE fueran comparables con aquellos de los pacientes recibiendo delandistrogene moxeparvovec. Estas características fueron seleccionadas al ser factores pronósticos importantes para esta población de pacientes. La cohorte CE utilizó grupos de datos contemporáneos tomados de estudios separados de la DMD, incluyendo ensayos clínicos aleatorizados controlados y estudios de historia natural tomados de 2016, con datos utilizados de la actualización de 2018 y 2021:
- FOR-DMD (NCT01603407): Un ensayo clínico histórico llevado a cabo de 2013 a 2019 para evaluar el tratamiento con corticosteroides en pacientes masculinos con DMD de 4 a < 8 años de edad. Se siguió a los pacientes por hasta 5 años (N=194). La última actualización de datos utilizada en este análisis fue en noviembre de 2021 [12].
- PRO-DMD-01 (NCT01753804): Un estudio de historia natural prospectivo de la progresión del daño físico, limitación de la actividad, y calidad de vida en la DMD se realizó de 2012 a 2016 en 16 sedes clínicas de 10 países en pacientes de 2.9 a 19.3 años. Se siguió a los pacientes por hasta 3 años (N=269) [13].
- CINRG DNHS (NCT00468832): Un estudio observacional multinacional realizado de 2006 a 2009 y de 2012 a 2016 en pacientes masculinos con DMD, de 2 a 28 años de edad. Se siguió a los pacientes por hasta 10 años (N=440). La última actualización de datos usada en este análisis fue el 18 de marzo de 2018 [14, 15].
Se aplicaron los criterios de inclusión y exclusión a la cohorte CE para asegurar consistencia y comparabilidad con las características observadas de los pacientes inscritos en el estudio EMBARK al momento de la administración del tratamiento. Los criterios de admisión para la cohorte CE fueron:
- Edad de 4 a < 8 años
- Puntaje total en la NSAA de ≥ 14 y ≤ 32 puntos
- Tiempo TTR ≤ 5.75 s
- Tiempo 10MWR ≤ 6.85 s
- Dosis estable de corticosteroides orales para ≥ 12 semanas
Esto creó un grupo de comparación consolidado y definido prospectivamente de pacientes con DMD recibiendo cuidado, emparejado con los pacientes tratados en la parte 1 del estudio EMBARK para variables como edad, sezo, uso de corticosteroides, puntaje total de inicio en la NSAA, pruebas de función cronometradas, estatura, peso, e índice de masa corporal. Se proporcionan más detalles sobre el uso de corticosteroides en la cohorte CE en la Tabla S1 del Material Suplementario. El análisis prospectivo definido por la puntuación de propensión permitió un balance robusto y riguroso de múltiples variables.
Análisis estadístico
El balance de las características de inicio entre el estudio EMBARK y las cohortes CE fue evaluado usando diferencias estandarizadas de medias (DME). Los cambios del punto de inicio en el puntaje total de la NSAA, TTR, tiempo 10MWR, velocidad para levantarse del suelo (RFF, por sus iniciales inglés), y velocidad de la medida 10MWR fueron analizados usando modelos de efectos mixtos para medidas repetidas (MMRM, por sus iniciales en inglés). El modelo incluyó un grupo de tratamiento (grupos EMBARK y CE), visita de análisis (año 1, año 2), interacción entre el grupo del tratamiento y la visita en el valor inicial, parámetro de valoración individual en el valor inicial, interacción del parámetro de valoración individual y la visita en el grupo de edad en el valor inicial, y la interacción entre el grupo de edad en el valor inicial y el parámetro de valoración individual en el valor inicial. También se incluyó el efecto del tratamiento en promedio en los pesos tratados obtenido en el modelo de puntaje de propensión. Los 64 pacientes tratados con delandistrogene moxeparvovec y los 143 pacientes en la cohorte CE fueron incluídos en los análisis; los métodos de medidas repetidas mediante un modelo mixto (MMRM) dan cuenta de los datos faltantes en estos análisis. Un paciente tratado con delandistrogene moxeparvovec en la parte 1 no tenía los datos de seguimiento de 2 años (la parte 2 de la semana 52 de la NSAA se consideró inválida por motivos de conducta). En la cohorte CE, hubo pacientes con evaluaciones del año 2 faltantes para la NSAA (n=9), TTR (n=28), y 10MWR (n=30). Todos los valores P son nominales y no han sido ajustados para comparaciones diferentes.
RESULTADOS
Características de inicio
En la parte 1 del ensayo EMBARK, 64 pacientes recibieron delandistrogene moxeparvovec: la edad promedio (rango) de inicio, 5.98 años (4.07-7.87); puntaje total en la NSAA, 23.3 (14-32) puntos. Ningún paciente perdió la habilidad de realizar evaluaciones funcionales durante el seguimiento de 2 años. Previo a la ponderación por puntaje de propensión, 155 pacientes en la cohorte EC (FOR-DMD, n=89; PRO-DMD-01, n=41; CINRG DNHS, n=25) cumplieron los criterios de inscripción pre-específicos con base en factores pronósticos clave (ver sección de Métodos) y tenían ≥ 1 visitas posteriores al inicio del estudio. Sólo esos pacientes en la cohorte CE recibiendo regímenes de corticosteroides de cuidado fueron incluídos en la ponderación por puntaje de propensión; la dosis promedio diaria para la cohorte CE es desconocida. Después de excluir a los pacientes con puntajes de propensión no superpuestas y ponderación del puntaje de propensión (ver sección de Métodos), la cohorte CE para el análisis pre-específico consistió de 143 pacientes: edad promedio (rango), 6.24 años (4.24-7.99); puntaje total en la NSAA, 23.5 (15-32) puntos. Las características de inicio fueron coincidentes entre delandistrogene moxeparvovec y la cohorte CE después de la ponderación del puntaje de propensión, evaluada por diferencias promedio estandarizadas (Tabla 1). Diferencias promedio estandarizadas para la edad y la estatura fueron >0.25; sin embargo, los valores promedio fueron similares entre los dos grupos.
Resultados funcionales
A los 2 años, los pacientes tratados con delandistrogene moxeparvovec (n=63; un paciente tratado en la parte 1 de EMBARK no tuvo datos de seguimiento de 2 años) demostraron beneficios funcionales estadísticamente significativos frente a la cohorte CE (Fig. 1 y 2) y resultados funcionales clave del cambio de inicio para el puntaje total en la NSAA (2.63 frente a – 0.25 puntos; LSM [95% CI] 2.88 [1.43–4.33]; P = 0.0001), TTR (0.65 frente a 2.71 s; − 2.06 [− 3.43 to − 0.70]; P = 0.0033), y 10MWR (− 0.04 frente a 1.32 s; − 1.36 [− 2.24 to − 0.47]; P = 0.0028); y los valores O son nominales y no han sido ajustados para múltiples comparaciones. El cambio del punto de inicio en la velocidad RFF (0.003 frente a − 0.053 aumento por segundo; 0.055 [0.03–0.08]; P < 0.0001) y 10MWR de velocidad (0.124 frente a − 0.115 m/s; 0.239 [0.12–0.36]; P = 0.0001) demostró también un beneficio funcional estadísticamente significativo frente a la cohorte CE (ver Figura S1 en la sección de Material Suplementario).
Otros parámetros funcionales fueron estables a los 2 años en pacientes tratados con delandistrogene moxeparvocec, aunque no había datos disponibles del CE para comparar. El cambio en promedio (desviación estándar) del punto de inicio fue – 0.03 (0.40) m/s en la velocidad de zancada en el centil 95, -2.30 (24.17) seg. en la prueba para caminar 10 metros, y 0.07 (3.51) seg. en el tiempo para subir cuatro escalones (Fig.3).
Para evaluar el impacto de corticosteroides adicionales en resultados funcionales, el puntaje total en la NSAA y otros parámetros funcionales fueron evaluados en pacientes que fueron tratados con delandistrogene moxeparvovec en la parte 1 y pasaron a la parte 2 desde la semana 52 a la semana 104 (Fig. 3, ver área sombreada). Estos pacientes recibieron una dosis de corticosteroides (1.6 mg./kg. al día de prednisona equivalente) desde el día -1 al día 60 en la semana 52 (cuando recibieron su parte 2 del tratamiento con un placebo salino). Aunque algunos pacientes recibieron un curso prolongado de corticosteroides para gestionar las enzimas del hígado elevadas al recibir delandistrogene moxeparvovec en la parte 1, todos los pacientes habían regresado a la dosis de inicio entre los días 60 y 84 posterior a la infusión. En general, los pacientes que recibieron delandistrogene moxeparvovec en la parte 1 recibieron una dosis promedio (rango) de corticosteroides de 0.80 (0.49-1.26) mg./kg. al día durante los primeros 2 años de este estudio. Como se muestra en la Fig. 3, los pacientes tratados con delandistrogene moxeparvovec en la parte 1 no tuvieron un beneficio funcional adicional aparente de los corticosteroides incrementados durante el paso al placebo en la parte 2.
Resultados biológicos
Los análisis realizados en las biopsias musculares en pacientes tratados en un subgrupo de sedes (preseleccionados según la experiencia en realizar biopsias musculares) a la semana 12 (n=17) y la semana 64 (n=16; un paciente fue transferido a una sede donde no se realizan biopsias en la parte 2 y una biopsia de la semana 64 no se tomó) demostraron expresión de microdistrofina mantenida y localización sarcolemal. El promedio (desviación estándar) del Western blot (% de control) fue de 34.29% (41.04%) a la semana 12 y 45.68% (30.75%) a la semana 64. El porcentaje de fibras positivas a distrofina fue de 28.13% (26.10%) a la semana 12 y 38.60% (26.93%) a la semana 64.
Seguridad
La seguridad desde el punto de inicio a la semana 52 se ha reportado previamente y es manejable con el monitoreo y tratamiento apropiado de los eventos adversos, los cuales ocurren normalmente dentro de los 90 días posteriores a la infusión. Entre las semanas 52 y 104 (n=63), hubieron 352 nuevos eventos adversos en 15 pacientes (23.81%): aumento de troponina-I (n= 4 pacientes, 6.35%), y la proteinuria y dolor de cabeza (n=2, 3.17% cada uno) fueron los más frecuentes (Fig. 4). Un paciente experimentó dos eventos adversos serios de rabdomiólisis relacionados al tratamiento; ambos eventos se resolvieron, uno con prednisolona administrada de forma oral e intravenosa, y el segundo sólo por vía intravenosa. Entre el punto de inicio y la semana 104, no se presentaron decesos relacionados al tratamiento, interrupciones del estudio debido a eventos adversos, o eventos adversos clínicamente significativos mediados por la activación del complemento. Fuera del ensayo EMBARK, a la fecha, han habido dos decesos relacionadas al tratamiento debido a insuficiencia hepática aguda aproximadamente 3 meses posteriores a la infusión en pacientes no ambulatorios (uno en el ensayo ENVISION [NCT05881408] y uno de marco comercial) a los 15 y 16 años de edad y en etapa avanzada de DMD [16].
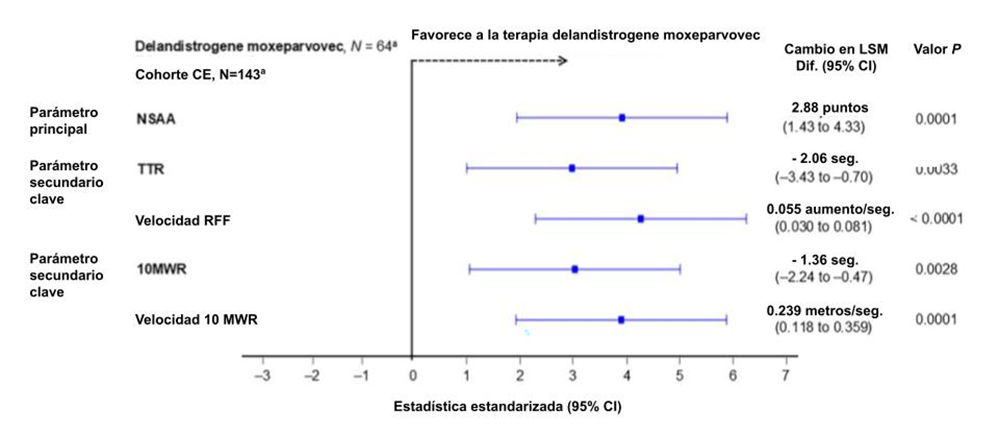
Fig. 1 Diagrama de bosque de los resultados funcionales de 2 años posteriores a la infusión de delandistrogene moxeparvovec comparada con la cohorte CE. a Todos los 64 pacientes tratados con delandistrogene moxeparvovec y los 143 pacientes en la cohorte CE fueron incluídos en los análisis; los métodos MMRM justifican los datos faltantes en estos análisis. Un paciente tratado con delandistrogene moxeparvovec en la parte 1 no presentó datos del seguimiento de 2 años (la escala NSAA de la parte 2 en la semana 52 se consideró inválida por razones de comportamiento). En la cohorte CE, 29, 28, y 30 pacientes tuvieron datos faltantes del año 2 para las evaluaciones de la NSAA, TTR, y 10MWR, respectivamente. Los valores negativos para las pruebas cronometradas funcionales (TTR, 10MWR) mostraron una mejora en el tiempo que toma lograr estos parámetros. Todos los valores P son nominales y no están ajustados para las diferentes comparaciones. Los métodos LSM (del cambio a partir del punto de inicio) y los CI fueron estandarizados al dividirlo entre el EE. Las diferencias del método LSM están en la escala original (sin el ajuste en el EE). Los signos de la función cronometrada fueron revertidos en el diagrama de bosque para alinear direcciones favorables entre los parámetros. Los resultados numéricos de la diferencia en el método LSM mantuvo los signos originales. Abreviaturas explicadas: 10MWR, prueba para caminar de 10 metros; CI, Intervalo de Confianza; CE, Control Externo; RFF, tiempo para levantarse del suelo; LSM, método de mínimos cuadrados; MMRM, modelos de efectos mixtos para medidas repetidas; NSAA, Escala Ambulatoria North Star; seg. segundos; EE, error estándar; TTR, tiempo para levantarse.
DISCUSIÓN
El tratamiento de la causa subyacente de la DMD, que usa terapia génica para restaurar la funcionalidad de la distrofina, tiene gran potencial para retrasar la progresión de la enfermedad; sin embargo, los beneficios a largo plazo y durabilidad no se han establecido aún. El estudio EMBARK, con los datos de 2 años de seguimiento, es el estudio clínico en curso más extenso y duradero sobre una terapia génica de microdistrofina para la DMD. Debido a que el brazo (grupo) del placebo se mantuvo sólo por un año, un CE se usó para evaluar el desempeño de pacientes frente al curso esperado de la enfermedad, definido por la historia natural. Para controlar la heterogeneidad de la DMD, se pueden usar métodos bien establecidos para hacer coincidir a los pacientes en la cohorte CE para la población del ensayo clínico, basados en los indicadores pronósticos del punto de inicio de la progresión de la enfermedad [8]. A pesar de su utilidad, las cohortes CE tienen limitaciones incluyendo el riesgo potencial del sesgo al seleccionar, la influencia de confundir las variables debido a la sola confianza en grupos de datos históricos, diferencias en las prácticas del manejo de la enfermedad, y mayor variabilidad inter-pacientes al punto de inicio; estos factores son controlados más efectivamente en un ensayo clínico aleatorizado. Los enfoques tales como los alineados con los criterios de admisión de la CE con las características de la población de pacientes en el ensayo clínico y aplicar la ponderación por puntajes de propensión pueden superar algunos de estos retos y facilitar la formación de una cohorte CE que esté emparejada apropiadamente con el grupo del tratamiento [8]. Estas limitaciones deberían considerarse cuando se interpretan los resultados, pero también ponderarse en proporción de la importancia de la evaluación continua de la eficacia a largo plazo.
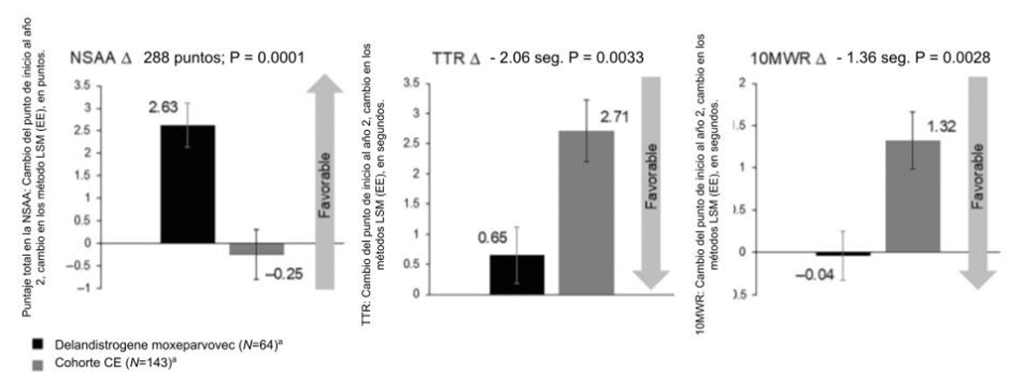
Fig. 2 Cambios del punto de inicio al año 2 en los métodos LSM, en resultados funcionales posteriores a la infusión de delandistrogene moxeparvovec en comparación con la cohorte CE. aTodos los 64 pacientes tratados con delandistrogene moxeparvovec y los 143 pacientes en la cohorte CE fueron incluídos en los análisis; los métodos MMRM justifican los datos faltantes en estos análisis. Un paciente tratado con delandistrogene moxeparvovec en la parte 1 no presentó datos del seguimiento de 2 años (la escala NSAA de la parte 2 en la semana 52 se consideró inválida por razones de comportamiento). En la cohorte CE, 29, 28, y 30 pacientes tuvieron datos faltantes del año 2 para las evaluaciones de la NSAA, TTR, y 10MWR, respectivamente. Los valores negativos para las pruebas cronometradas funcionales (TTR, 10MWR) mostraron una mejora en el tiempo que toma lograr estos parámetros. Todos los valores P son nominales y no están ajustados para las diferentes comparaciones. Abreviaturas explicadas: 10MWR, prueba para caminar de 10 metros; CI, Intervalo de Confianza; CE, Control Externo; RFF, tiempo para levantarse del suelo; LSM, método de mínimos cuadrados; MMRM, modelos de efectos mixtos para medidas repetidas; NSAA, Escala Ambulatoria North Star; seg. segundos; EE, error estándar; TTR, tiempo para levantarse.
La cohorte CE utilizada para el ensayo EMBARK se derivó de los estudios clínico (FOR-DMD) y observacional (PRO-DMD-01, CINRG DNHS) y estadísticamente ponderados para emparejarse con las características de los pacientes tratados con delandistrogene moxeparvovec en la parte 1 del estudio EMBARK. A los 6 a 10 años de edad (aproximadamente 2 años posteriores a la infusión en la población del estudio EMBARK [tratados con delandistrogene moxeparvovec de los 4 a < 8 años de edad]), los pacientes con DMD están cambiándose típicamente de la fase plateau en la NSAA (habiendo alcanzado el pico en la función motora) en la fase de declive funcional temprana [17]. Aunque no se espera que la terapia génica de microdistrofina revierta el daño muscular preexistente, el tratamiento con delandistrogene moxeparvovec en la población del estudio EMBARK (en la fase de adquisición de habilidades o plateau de la NSAA al inicio del estudio) podría haber permitido a los pacientes alcanzar un pico de desempeño al estabilizar o retrasar la degeneración muscular subyacente. Los resultados del estudio EMBARK de 2 años muestran la habilidad funcional estabilizada que se mantuvo del punto de inicio a los 2 años, en comparación con la cohorte CE emparejada descendente, sugiriendo que los pacientes en el estudio EMBARK habrían descendido por debajo de su punto de inicio durante los 2 años sin tratamiento. Los resultados funcionales del TTR y 10MWR, pronóstico de la pérdida de ambulación retrasada, también demostraron estabilización o retraso de la progresión de la enfermedad en el grupo tratado en comparación con la cohorte CE a los 2 años [17–19]. Si la terapia delandistrogene moxeparvovec continúa retrasando la progresión de la enfermedad, se anticipó una gran divergencia de la historia natural con el seguimiento a largo plazo conforme los pacientes en la cohorte en la CE continúan en la fase de declive de la enfermedad.
Un factor potencialmente complicado es que los pacientes en el estudio EMBARK recibieron una dosis aumentada de corticoesteroides en el período de peri-infusión, al inicio de las partes 1 y 2. Esta dosis de esteroides aumentada fue establecida por el protocolo del estudio para ayudar a mitigar los eventos adversos inmunomediados en respuesta a la administración de vectores. Los corticosteroides han documentado beneficios al mejorar los resultados motores en pacientes con DMD [20], y pacientes recibiendo delandistrogene moxeparvovec en el estudio EMBARK recibieron una dosis de corticosteroide promedio de 0.80 (rango 0.49-1.26) mg./kg. al día durante los primeros 2 años del estudio. Aunque el promedio de dosis diaria de corticosteroides para la cohorte CE es desconocida, es posible que los pacientes en el estudio EMBARK recibieron niveles más altos de corticosteroides en promedio relativos a los comparadores de la CE. Una opinión longitudinal del puntaje total en la NSAA y otros resultados funcionales de pacientes tratados con delandistrogene moxeparvovec en la parte 1 podrían ser útiles al considerar el impacto de los esteroides peri-infusión (Fig. 3). De manera notable, los 61 días de corticosteroides aumentados a la semana 52 junto con la infusión del placebo no tuvo efecto clínicamente significativo en los puntajes totales en la NSAA u otras medidas funcionales. Los resultados de dos años deberían interpretarse con esta información y limitaciones potenciales en mente. La evaluación a largo plazo continua de los pacientes tratados en la parte 1 del estudio EMBARK permitirá mayor evaluación de los resultados funcionales con la separación del periodo de dosis peri-infusión de corticosteroides y podría ofrecer una clara perspectiva del impacto en los resultados a largo plazo.
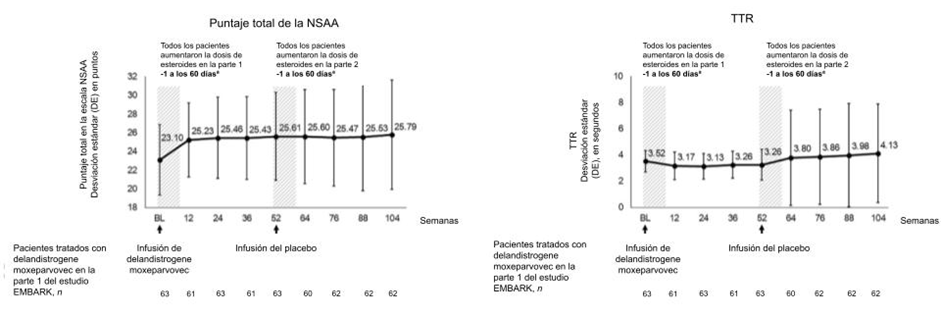
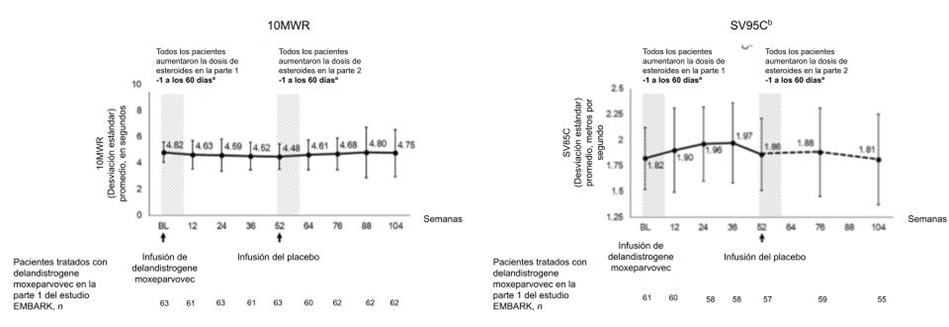
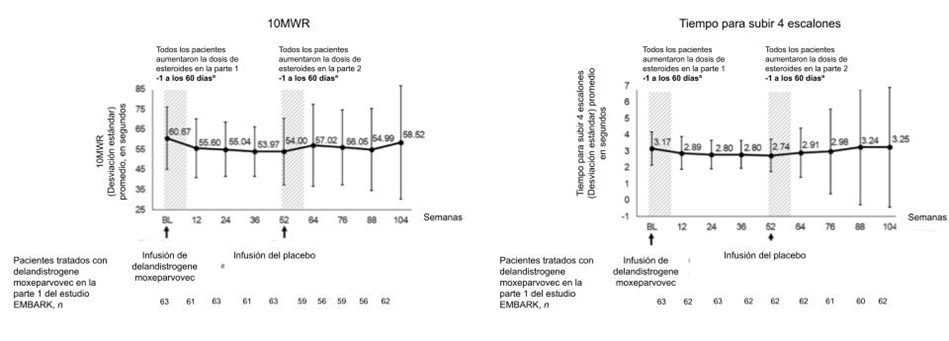
Fig. 3 Resultados funcionales de dos años en pacientes tratados con delandistrogene moxeparvovec la parte 1 del estudio EMBARK. aLa dosis de corticosteroides fue aumentada en la parte 1 y parte 2 desde el día -1 al 60. bLos datos no están disponibles para las semanas 64 y 88 (las líneas discontinuas cubren los puntos temporales sin valores). Abreviaturas explicadas: 10MWR, prueba para caminar de 10 metros; 100MWR, prueba para caminar de 100 metros; seg., segundos; DE, desviación estándar; SV95C, velocidad de zancada en el centil 95, SV95C; TTR, tiempo para levantarse del suelo.
La expresión de microdistrofina mantenida y la localización en el sarcolema se observaron en las biopsias de la semana 64. Estos resultados mantenidos sugieren el potencial de delandistrogene moxeparvovec de abordar de forma persistente la causa subyacente de la DMD y ayudar a retrasar el daño muscular por un periodo de tiempo prolongado.
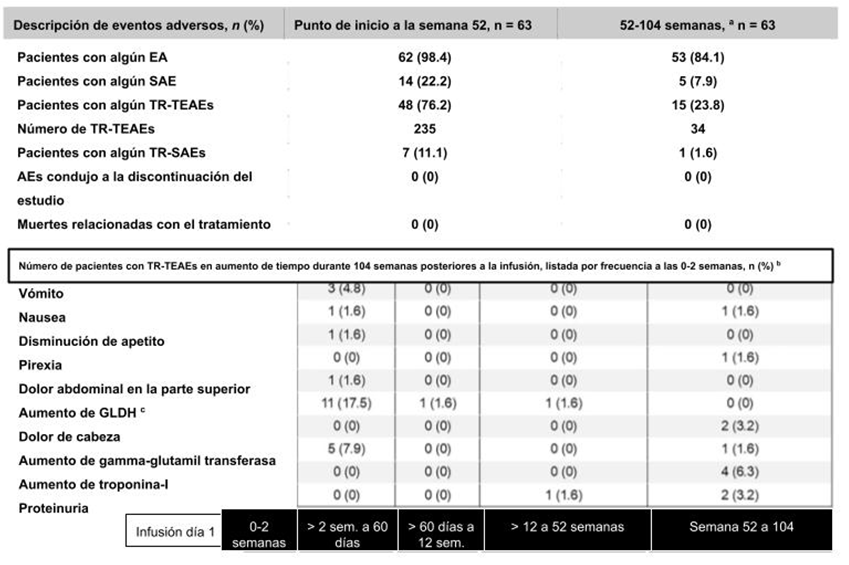
Fig. 4 Descripción de la seguridad del estudio EMBARK. a Nuevos eventos entre la semana 52 y 104 (excluyendo eventos en curso que comenzaron durante la parte 1 [del punto de inicio a la semana 52]. b TR-TEAEs en menos del 10% de los pacientes en la parte 1 o menos del 3% de los pacientes en la parte 2. c Basado en evaluaciones de un investigador y el rango normal de la institución. Abreviaturas explicadas: EA, eventos adversos; GLDH, glutamato deshidrogenasa; SAE, eventos adversos serios; TEAE, eventos adversos emergentes del tratamiento; TR-SAE, evento adverso serio relacionado al tratamiento; TR-TEAE, evento adverso emergente del tratamiento, relacionado al tratamiento.
Además de las limitaciones de la cohorte CE descritos anteriormente, una limitación del estudio clave es la pérdida de un brazo (grupo) del placebo después de 1 año; como resultado de la naturaleza irreversible y progresiva de la DMD, no redunda en el interés de los pacientes mantener los grupos de placebo durante períodos prolongados [21].
CONCLUSIÓN
A los 2 años, delandistrogene moxeparvovec demostró estabilización o retraso de la progresión de la enfermedad de la DMD frente a la cohorte CE emparejada. Los resultados de seguridad de dos años fueron consistentes con los resultados de seguridad de 1 año del estudio EMBARK, sin signos nuevos de seguridad observados entre la semana 52 y la semana 104 [5–7, 22].
AGRADECIMIENTOS
Agradecemos a los pacientes que participaron en este ensayo, sus familias y cuidadores; los miembros del comité de monitoreo de datos; el personal que asistió con el ensayo en cada sede, al grupo del estudio EMBARK; los equipos patrocinadores del estudio que respaldaron este ensayo; los estudios FOR-DMD, PRO-DMD-01, y CINRG DNHS para los datos externos cohorte; y Cindy Chen y Bagirathy Ravishankar de Sarepta Therapeutics, Inc. y Anne Wong de F. Hoffmann-La Roche Ltd por el apoyo en el desarrollo del manuscrito. El grupo del estudio EMBARK y el investigador, y sedes de reclutamiento han sido publicados previamente en Mendell JR, et al, Nat Med. 2025; 31:332-341.
Escritura Médica, Editorial y Otra Asistencia. La escritura médica y asistencia editorial se proporcionaron por Emily Turner y Nucleus Global de acuerdo con los lineamientos de 2022 de las Buenas Prácticas de Publicación (GPP, por sus siglas en inglés) (https://www.ismpp.org/gpp-2022) financiadas por Sarepta Therapeutics, Inc. y F. Hoffman-La Roche Ltd.
Contribuciones de Autores. Jerry R. Mendell, Francesco Muntoni, Eugenio M. Mercuri, Hirofumi Komaki, y Ulrike Schara‑Schmidt conceptualizaron o diseñaron el trabajo, datos adquiridos; datos interpretados; esbozaron el trabajo o lo revisaron críticamente; aprobaron la versión enviada; y estuvieron de acuerdo con ser responsables por la precisión e integridad de todos los aspectos del trabajo. Craig M. McDonald adquirió datos; analizó datos; interpretó datos; esbozó el trabajo o lo revisó críticamente; aprobó la versión enviada; y estuvo de acuerdo con ser responsables por la precisión e integridad de todos los aspectos del trabajo. Emma Ciafaloni, Carmen Leon-Astudillo, Andrés Nacimiento; Crystal Proud; Aravindham Veerapandiyan, y Craig M. Zaidman adquirieron datos; interpretaron datos; esbozaron el trabajo o lo revisaron críticamente; aprobaron la versión enviada; y estuvieron de acuerdo con ser responsables por la precisión e integridad de todos los aspectos del trabajo. Matthew Furgerson, Kai Ding, Preeti Singh, Rachael Potter, Damon R. Asher, Alexander P. Murphy, Carol Reid, Gregory Hooper, Carmen O. Torre, y Marianna Manfrini analizaron datos; interpretaron datos; esbozaron el trabajo o lo revisaron críticamente; aprobaron la versión enviada; y estuvieron de acuerdo con ser responsables por la precisión e integridad de todos los aspectos del trabajo. Louise R. Rodino-Klapac conceptualizó o diseñó el trabajo; analizó datos; interpretó datos; esbozó el trabajo o lo revisó críticamente; aprobó la versión enviada; y estuvo de acuerdo con ser responsables por la precisión e integridad de todos los aspectos del trabajo. Todos los coautores han revisado y aprobado los contenidos de este manuscrito, y todos los requisitos de autoría se han cumplido. De acuerdo con los criterios del International Committee of Medical Journal Editors (ICMJE). Todos los autores contribuyeron al diseño del estudio, recolección de datos, análisis, interpretación, escritura del manuscrito, revisión, y aprobación, y decidieron conjuntamente publicar el manuscrito.
Financiación. Este ensayo fue patrocinado por Sarepta Therapeutics, Inc., y financiado por Sarepta Therapeutics, Inc. y F. Hoffmann-La Roche Ltd. El patrocinador tuvo la responsabilidad final del diseño, protocolo, mantenimiento de la base de datos, realización del ensayo, análisis de datos, y confirmación de datos en el ensayo. La tarifa por revisión acelerada de la revista fue financiada por los patrocinadores del estudio (Sarepta Therapeutics, Inc. y F. Hoffmann-La Roche Ltd.).
Disponibilidad de datos. Los grupos de datos generados durante y/o analizados durante el presente estudio están disponibles previa solicitud al autor correspondiente. Investigadores calificados pueden solicitar a Sarepta, Therapeutics, Inc. acceso a los datos que respalden los hallazgos de este estudio poniéndose en contacto al siguiente correo electrónico: medinfo@sarepta.com, sujeto a previa revisión de los patrocinadores del estudio, con base en un criterio de caso por caso.
Declaraciones
Conflictos de interés. La afiliación de Jerry R. Mendell al momento de la parte 1 del estudio EMBARK fue el Centro para Terapia Génica, del Hospital Infantil Nacional, Columbus, Ohio, EE. UU. (actualmente empleado por Sarepta Therapeutics, Inc.); la afiliación de Jerry R. Mendel al momento de la parte 2 del estudio EMBARK y afiliación actual es Sarepta Therapeutics, Inc., Cambridge, MA, EE. UU. Jerry R. Mendel recibió la financiación del estudio de Sarepta Therapeutics, Inc. mientras fue en el Hospital Infantil Nacional al momento del estudio y es actualmente un empleado de Sarepta Therapeutics, Inc. Jerry R. Mendell es co-inventor de la tecnología AAVr h74.MHC 7.micro-dys. Francesco Muntonu ha recibido honores y subsidios de Sarepta Therapeutics, Inc. por participar en simposios y comités asesores, y forma parte como investigador en Sarepta Therapeutics, Inc. Reporta participación en comités asesores de Novartis, F. Hoffmann-La Roche Ltd, Edgewise Therapeutics , Dyne Therapeutics, Pfizer, PTC Therapeutics, e Italfarmaco. Craig M. McDonald reporta subsidios de Capricor Therapeutics, Catabasis, Edgewise Therapeutics, Epirium Bio, Italfarmaco, Pfizer, PTC Therapeutics, Santhera Pharmaceuticals, y Sarepta Therapeutics, Inc. y tiene un rol con Biomarin, Capricor Therapeutics, Catalyst, Edgewise Therapeutics, Italfarmaco, PTC Therapeutics, F. Hoffman-La Roche Ltd., Santhera Pharmaceuticals y Sarepta Therapeutics, Inc. Ha recibido honores de PTC Therapeutics y Sarepta Therapeutics, Inc. Eugenio M. Mercuri ha recibido cuotas de AveXis, Biogen, y F. Hoffman-La Roche Ltd. Emma Ciafaloni ha recibido honores de Sarepta Therapeutics, Inc. por participar en comités asesores e investigación y/o apoyo de subsidios de Centros para el Control y Prevención de Enfermedades, CuareSMA, la asociación Muscular Dystrophy Association, los National Institutes of Health, Orphazyme, el Instituto de Investigación de Resultados Centrados en el Paciente, Parent Project Muscular Dystrophy, PTC Therapeutics, Inc., y la Administración de Alimentos y Medicamentos de EE. UU. Hirofumi Komaki ha recibido subsidios de Sarepta Therapeutics, Taiho Pharmaceutical Co., Ltd., Chugai Pharmaceutical Co., Nippon Shinyaku Co., Ltd., y Kaneka Corporation. Hirofumi Komaki ha recibido cuotas de Sarepta Therapeutics, Inc., Pfizer, PTC Therapeutics, Chugai Pharmaceutical Co., Nippon Shinyaku Co., y Kaneka Corporation. Carmen Leon-Astudillo es investigadora en los ensayos clínicos de Sarepta Therapeutics, Inc. y sub-investigadora en estudios patrocinados por Pfizer, SolidBiosciences, Edgewise Therapeutics, Italfarmaco, y Genentech/Roche. Andrés Nascimento ha recibido cuotas de AveXis, Biogen, y F. Hoffman-La Roche Ltd. Crystal Proud participa en un comité asesor y es consultor para Biogen, Sarepta Therapeutics, Inc., AveXis/Novartis Gene Therapies, Genentech/Roche, y Scholar Rock; sirve como orador para Biogen; y es investigador principal de estudios patrocinadores por AveXis/Novartis Gene Therapies, AMO Pharma, Astellas, Biogen, CSL Behring, Fibrogen, PTC Therapeutics, Pfizer, Sarepta Therapeutics, Inc., y Scholar Rock. Urike Schara-Schmidt ha recibido honorarios por asesoramiento y participación en conferencias invitadas de Sarepta Therapeutics, Inc., y F. Hoffmann-La Roche Ltd. Aravidhan Veerapandiyan tiene un rol de consultoría/consejería con AMO Pharma, AveXis, Biogen, Edgewise Therapeutics, FibroGen, Novartis, Pfizer, PTC Therapeutics, Inc., UCB Pharma, Catalys, y Scholar Rock; ha recibido financiamiento de investigación de AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, la asociación Muscular Dystrophy Association, Novartis, Parent Project Muscular Dystrophy, Pfizer, RegenBio, y Sarepta Therapeutics, Inc., y tiene otras relaciones con MedLink Neurology para servicios editoriales. Craig M. Zaidman ha recibido apoyo en investigaciones de Biogen y Novartis y ha servido en un comité asesor para Sarepta Therapeutics, Inc. Matthew Furgerson, Kai Ding, Preeti Singh, Rachel Potter, y Damon R. Asher son empleados de Sarepta Therapeutics, Inc. y puede tener opciones de compra de acciones. La afiliación de Kai Ding al momento de la Parte 1 del ensayo EMBARK fue Sarepta Therapeutics, Inc., Cambridge Ma, EE. UU.; Kai Ding ya no trabaja para Sarepta Therapeutics. Alexander P. Murphy, Carol Reid, Gregory Hooper y Carmen O. Torre son empleados de Roche Products Ltd y pueden tener opciones de compra de acciones en F. Hoffmann-La Roche Ltd. Marianna Manfrini trabaja para F. Hoffmann-La Roche Ltd y puede tener opciones de compra de acciones. Louise R. Rodino-Klapac es empleada de Sarepta Therapeutics, Inc. y puede tener opciones de compra de acciones. Además, es co-inventora de la tecnología AAVr h74.MHC 7.micro-dys.
Aprobación ética. El diseño del estudio EMBARK, incluyendo la selección de pacientes, declaraciones de ética (incluyendo una lista completa de comités revisores institucionales y comités de ética), y descripciones de terapia génica, han sido publicados previamente por completo [7]. Este ensayo fue realizado en cumplimiento con los lineamientos de Buenas Prácticas y la Declaración de Helsinki. El comité de revisión institucional y el comité de ética en cada sede aprobó el protocolo del ensayo y todas sus modificaciones. Los detalles de todos los comités de revisión institucionales y comités de ética están disponibles en el Material Suplementario. El protocolo del ensayo está disponible bajo solicitud del autor correspondiente. El análisis principal del estudio se llevó a cabo después de que todos los pacientes completaran la parte 1 y estos hallazgos han sido publicados [7]. Todos los autores estuvieron involucrados en el diseño del estudio, la colección de datos, análisis, e interpretación, así como la redacción, revisión, y aprobación del manuscrito y acordaron en conjunto su publicación [7]. El patrocinador fue responsable de confirmar el diseño, protocolo, y realización del ensayo final, junto con la base de datos, mantenimiento, análisis de datos, y precisión de los datos [7]. Todos los autores recopilaron y tuvieron acceso completo a los datos, y atestiguaron a su precisión y totalidad de acuerdo con el protocolo del ensayo [7]. Los padres de familia o tutores legales ofrecieron consentimiento informado y, cuando fuera aplicable, se obtuvo el consentimiento de los pacientes. La seguridad, eficacia, calidad de los datos, e integridad del estudio continúan siendo monitoreados por un comité independiente de monitoreo de datos [7].
Acceso Abierto. Este artículo tiene una licencia de Creative Commons de Reconocimiento No comercial 4.0 Internacional, la cual permite cualquier uso no comercial, distribución, adaptación, distribución, y reproducción en cualquier medio o formato, siempre y cuando le dé crédito apropiado al autor(es) original(es) y la fuente, dar el enlace a la licencia Creative Commons, e indicar si realizaron cambios. Las imágenes u otro material de terceras partes en este artículo están incluídos en la licencia Creative Commons del artículo, a menos que se indique lo contrario en una línea otorgando el crédito en el material. Si el material no está incluído en la licencia Creative Commons del artículo y el uso que se pretende dar no está permitido por regulación estatutaria o excede el uso permitido, necesitará obtener permiso directamente del propietario de copyright. Para revisar una copia de esta licencia, visite el enlace: http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCIAS
1. Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7:13.
2. Bengtsson NE, Tasfaout H, Chamberlain JS. The road towards AAV-mediated gene therapy
of Duchenne muscular dystrophy. Mol Ther. 2025;33:2035–51.
3. US Food and Drug Administration. ELEVIDYS® Highlights of prescribing information. 2025.
https://www.elevidyshcp.com/pi. Accessed December 2025.
4. Mendell J, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.microdystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020; 77:1121–31.
5. Mendell JR, Shieh PB, McDonald CM, et al. Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front Cell Dev Biol. 2023;11:1167762.
6. Zaidman CM, Proud CM, McDonald CM, et al. Delandistrogene moxeparvovec gene therapy in ambulatory patients (aged ≥4 to <8 years) with Duchenne muscular dystrophy: 1-year interim
results from study SRP-9001-103 (ENDEAVOR). Ann Neurol. 2023;94:955–68.
7. Mendell JR, Muntoni F, McDonald C, et al. AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial. Nat Med. 2024;31:332–41.
8. Muntoni F, Signorovitch J, Sajeev G, et al. Realworld and natural history data for drug evaluation in Duchenne muscular dystrophy: suitability of the North Star Ambulatory Assessment for comparisons with external controls. Neuromuscul Discord. 2022;32:271–83.
9. Goemans N, Signorovitch J, Sajeev G, et al. Suitability of external controls for drug evaluation
in Duchenne muscular dystrophy. Neurology. 2020;95:e1381–91.
10. US Food & Drug Administration. Considerations for the design and conduct of externally controlled trials for drug and biological prod ucts. 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-and-conduct-externally-controlled-trials-drug-and-biological-products. Accessed October 2025.
11. Rubin DB. Using propensity scores to help design observational studies: application to the tobacco litigation. Health Serv Outcomes Res Methodol. 2001;2:169–88.
12. ClinicalTrials.gov. NCT01603407: Finding the optimum regimen for Duchenne muscular dystrophy (FOR-DMD). Bethesda, MD: National Library of Medicine (US). https://www.clinicaltrials.govct2/show/NCT01603407. Accessed March 2025.
13. ClinicalTrials.gov. NCT01753804: A prospective natural history study of progression of subjects with Duchenne muscular dystrophy. 2025. https://clini caltrials.gov/study/NCT01753804?intr=NCT01753804&rank=1. Accessed March 2025.
14. ClinicalTrials.gov. NCT00468832: Longitudinal study of the natural history of Duchenne muscular dystrophy (DMD). 2016. https://clinicaltrials.gov/ct2/show/NCT00468832. Accessed March 2025.
15. Spurney C, Shimizu R, Morgenroth LP, Kolski H, Gordish-Dressman H, Clemens PR. Cooperative International Neuromuscular Research Group Duchenne Natural History Study demonstrates insufficient diagnosis and treatment of cardiomyopathy in Duchenne muscular dystrophy. Muscle Nerve. 2014;50:250–6.
16. Mendell JR, Muntoni F, McDonald CM, et al. Delandistrogene moxeparvovec in Duchenne muscular dystrophy: long-term EMBARK 2-year functional outcomes, safety, and micro-dystrophin expression. The 30th Annual International Congress of the World Muscle Society (WMS); 2025 October 7–11, 2025; Vienna, Austria.
17. Zambon AA, Ayyar Gupta V, Ridout D, et al. Peak functional ability and age at loss of ambulation in Duchenne muscular dystrophy. Dev Med Child Neurol. 2022;64:979–88.
18. Mazzone ES, Coratti G, Sormani MP, et al. Timed rise from floor as a predictor of disease progression in Duchenne muscular dystrophy: an observational study. PLoS ONE. 2016;11:e0151445.
19. McDonald CM, Signorovitch J, Mercuri E, et al. Functional trajectories before and after loss of ambulation in Duchenne muscular dystrophy and implications for clinical trials. PLoS ONE. 2024;19:e0304099.
20. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular
dystrophy, Part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–67.
21. Mercuri E, Asher D, Ding K, et al. Analytical framework for assessment of long-term efficacy of therapies for Duchenne Muscular dystrophy using external controls. Muscular Dystrophy Association (MDA) Clinical and Scientific Conference; 16–19 March; Dallas, TX, USA 2025. Available from: https://www.mdaconference.org/abstract-library/analytical-framework-for-assessment-of-long-term-efficacy-of-therapies-for-duchenne-muscular-dystrophy-using-external-controls/.
22. Mendell JR, Sahenk Z, Lehman KJ, et al. Long-term safety and functional outcomes of delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy: a phase 1/2a nonrandomized trial. Muscle Nerve. 2024;69:93–8.
Esta publicación está escrita en español para promover el acceso de la comunidad a información y actualizaciones. Las tablas y gráficas contenidas aquí fueron tomadas de la original y editadas con el texto traducido a español para promover el acceso de la comunidad a información. La información que se comparte tiene como objetivo el interés y la concientización general y, por lo tanto, no debe ser considerada ni interpretada como consejo médico o legal. Se trata de información publicada abiertamente. La Fundación Akari no promociona ninguna empresa, producto o tratamiento específico. Le recomendamos que consulte la fuente original y tome decisiones informadas.
Para leer el artículo de investigación original en inglés consulte:
Si desea saber más información acerca de este artículo, contáctese directamente con la fuente original del mismo.