Síguenos en Nuestras Redes Sociales
3 de noviembre de 2025 4:05 PM Hora Estándar del Este
- Sarepta anuncia la completación de su compromiso del ensayo confirmatorio para sus terapias de morfolino fosforodiamidato (PMOs, por sus iniciales en inglés) AMONDYS 45 y VYONDYS 53 para las enfermedades muy raras:
- Aunque el estudio ESSENCE no obtuvo una significancia estadística en su parámetro principal, los resultados indican tendencias positivas y motivadoras a favor de la terapia a las 96 semanas
- Sarepta reporta que el estudio estuvo impactado por la pandemia por COVID-19 y, cuando los datos impactados por COVID fueron excluídos, el efecto del tratamiento significativo se observa en el parámetro principal
- El estudio ESSENCE respaldó el perfil de seguridad favorable de las terapias AMONDYS 45 y VYONDYS 53
- La compañía espera agendar una reunión con la Administración de Alimentos y Medicamentos de EE. UU. (FDA, por sus siglas en inglés) y dialogar sobre una opción para la aprobación tradicional basada en el perfil riesgo-beneficio positivo de las terapias derivado de los resultados del estudio ESSENCE, así como de evidencia positiva significativa de varios años y del mundo real
- Los ingresos netos por productos para el tercer trimestre de 2025 dieron un total de $370.0 millones, los cuales consistieron en $238.5 M de PMOs y $131.5 M de ELEVIDYS
- El refinanciamiento de una porción mayoritaria de las Notas e iniciativas de costos por reestructuración de 2027 fortalecieron la posición financiera general
- Hay progreso en las conversaciones en cuanto al etiquetado de ELEVIDYS y se espera que concluyan pronto
- Las presentaciones en el congreso de la World Muscle Society, edición 2025, se agregaron al cuerpo de evidencias en relación a la eficacia y seguridad de ELEVIDYS
CAMBRDGE, Massachusetts. – (BUSINESS WIRE) – Sarepta Therapeutics, Inc. (NASDAQ:SRPT), el líder en medicina genética de precisión para las enfermedades raras, reportó hoy los resultados financieros del tercer trimestre de 2025 y la completación de ESSENCE, su estudio aleatorizado de Fase 3, doble ciego, controlado con placebo, global, que evalúa la eficacia y seguridad de AMONDYS 45 (casimersen) y VYONDYS 53 (golodirsen) en comparación con el placebo en 225 pacientes de 6 a 13 años de edad, con distrofia muscular de Duchenne (Duchenne) susceptibles a la omisión del exón 45 ó 53.
Resultados principales del estudio ESSENCE
Los resultados principales que se encontraron en las tendencias numéricas favorecieron el tratamiento frente al placebo. Sin embargo, la diferencia observada de 0.05 pasos por segundo en el método de mínimos cuadrados (LSM, por sus iniciales en inglés), no alcanzaron significancia estadística (P=0.309) en el parámetro principal, la velocidad de ascenso en 4 pasos (4SC, por sus iniciales en inglés) a las 96 semanas.
El estudio ASSENCE se llevó a cabo durante un periodo de nueve años que incluyó la pandemia por COVID-19, lo cual impactó a los participantes y los resultados del estudio. Un análisis que excluye datos de los participantes cuyo periodo doble ciego coincidió con la pandemia por COVID-19 (n=57), muestra una reducción del 30% (LSM 0.11 pasos por segundo, P=0.09) en la progresión de la enfermedad por 2 años en la 4SC en los participantes tratados que no fueron afectados por COVID frente al placebo (n=168). Esto representa un cambio clínicamente significativo.
No hubo nuevas señales de seguridad en el estudio ESSENCE, reforzando el perfil de seguridad favorable y estable observado con las terapias de omisión de exón a lo largo de los años. Los eventos adversos fueron leves en su mayoría (88%) o moderados (10.3%) y comparables entre el tratamiento y los grupos de placebo. Los eventos adversos emergentes del tratamiento más comunes (≥10%) fueron vómito, nasofaringitis, pirexia, dolor de cabeza, tos, caídas, e infecciones del tracto respiratorio superior.
Por más de una década, las terapias de morfolino fosforodiamidato (PMOs, por sus iniciales en inglés) de Sarepta se han usado para tratar poco más de 1,800 pacientes a nivel mundial, desde menores de hasta 7 meses de edad hasta adultos en sus 30 años. Los resultados de ESSENCE complementan la evidencia disponible de VYONDYS 53 y AMONDYS 45, incluyendo estudios del mundo real que demuestran, por ejemplo, que el tratamiento con VYONDYS 53 está asociado con un retraso de 7.5 años en la necesidad de usar ventilación durante la noche1 y el tratamiento con AMONDYS 45 está relacionado con una ralentización estadísticamente significativa del declive de la función pulmonar y un posible beneficio significativo en el tiempo previsto para usar un dispositivo de asistencia para la tos2. A través de nuestro portafolio de PMO, la evidencia del mundo real indica un beneficio de varios años en la mortalidad3,4, retratos en el tiempo para la pérdida de ambulación de 3 y 4 años5,6, una reducción sustancial en el riesgo de alcanzar una fracción de eyección ventricular izquierda (LVEF, por sus iniciales en inglés) de menos del 55%7, y una reducción importante en las visitas al área de emergencias y otras visitas al hospital8.
Basados en las tendencias prometedoras observadas en el estudio ESSENCE, la evidencia sustancial del mundo real, y el perfil de seguridad positivo de AMONDYS 45 y VYONDYS 53, Sarepta pretende agendar una reunión con la Administración de Alimentos y Medicamentos de EE. UU. (FDA, por sus siglas en inglés) para hablar sobre la posibilidad de pasar de una aprobación acelerada a una tradicional.
Sarepta continúa analizando los resultados del estudio ESSENCE y, junto con la evidencia del mundo real, los resultados completos serán enviados a la FDA como parte del llenado de una solicitud complementaria de nuevo fármaco (sNDA, por sus siglas en inglés) para estas dos terapias de omisión de exón. Se espera que la finalización del estudio ESSENCE satisfaga el requerimiento primario posterior a la comercialización. Además, los resultados del estudio ESSENCE se compartirán en reuniones médicas futuras, y se buscará la publicación en una revista médica.
“Aunque el estudio ESSENCE no logró la importancia estadística de su parámetro principal, creemos que los resultados demostraron un efecto del tratamiento clara, mostrando resultados funcionales clínicamente significativos para las personas con Duchenne que tienen mutaciones susceptibles a la omisión del exón 45 ó 53. Estos hallazgos principales refuerzan el posible impacto de estas terapias para ralentizar la debilidad muscular y otros síntomas. Los resultados del estudio son consistentes con el cuerpo creciente de evidencia del mundo real acumulada a lo largo de los años. Estos datos, los cuales hemos compartido con la FDA, fortalecen nuestra confianza en el beneficio de la distrofina añadida con el tiempo,” dijo Louise Rodino-Klapac, Ph.D. y presidenta del área de investigación y desarrollo, y operaciones técnicas de Sarepta.
La Dra. Rodino-Klapac continuó: “Este ensayo inscribió un subgrupo muy raro de pacientes Duchenne que eran elegibles. La complejidad de Duchenne, combinados con la heterogeneidad de la población y el impacto de la pandemia por COVID, hicieron de esto un proyecto extraordinario. Estamos profundamente agradecidos con las familias e investigadores cuya dedicación hizo esto posible, y nos mantenemos comprometidos con avanzar en el cuidado de la comunidad Duchenne al ofrecer opciones que pueden cambiar el curso de la distrofia muscular de Duchenne.
“En el ensayo y en mi práctica clínica, he dado seguimiento a niños y hombres jóvenes tratados con casimersen y golodirsen desde sus aprobaciones iniciales y, en mi opinión, estas terapias pueden ayudar a preservar funciones fundamentales como caminar, subir escalones y comer por sí mismos. Con el tiempo, estos beneficios pueden traducirse en una pérdida de la ambulación retrasada y un declive respiratorio incluso más lento, ofreciendo potencialmente a estas personas una opción significativa para mantener la calidad de vida,” dijo Craig McDonald, Doctor in Medicine, profesor y presidente del Departamento de Medicina Física y Rehabilitación de UC Davis Health, así como investigador en el estudio ESSENCE.
Puntos sobresalientes para el trimestre que terminó el 30 de septiembre de 2025
“Estamos muy complacidos de haber cumplido con nuestra obligación principal posterior a la comercialización con la completación del estudio ESSENCE, un ensayo particularmente retador de ejecutar en el contexto de estas enfermedades muy raras que causan la degeneración de manera heterogénea con el curso de las décadas. Esperamos comentar los resultados de ESSENCE y la evidencia del mundo real de AMONDYS 45 y VYONDYS 53 con la FDA,” según dijo Doug Ingram, director ejecutivo de Sarepta.
Continuó diciendo Ingram que “estamos también complacidos de reportar un desempeño sólido en el trimestre de nuestra terapia génica, ELEVIDYS, y nuestros tres PMO: EXONDYS 51, VYONDYS 53, Y AMONDYS 45. Nuestros ingresos netos por productos se mantuvieron en $370.0 millones para el trimestre. Además, el haber tomado los pasos para fortalecer nuestra posición económica, incluyendo el refinanciamiento de nuestra deuda convertible y una reestructuración financiera significativa, me complace reportar un flujo de dinero positivo para el trimestre. Viendo hacia adelante, tenemos una posición económica fuerte de la cual continuar sirviendo a nuestra comunidad conforme avanzamos en un portafolio de ARN pequeño de interferencia (ARNpi).”
- ELEVIDYS (delandistrogene moxeparvovec-rokl) Diálogos regulatorios: Se espera que las conversaciones entre la FDA y Sarepta sobre el proceso de etiquetado hayan finalizado en el futuro cercano con un resultado que incluya una advertencia en la caja y la eliminación de la indicación no ambulatoria sobre la sección ‘Indicaciones y usos’ en la información de receta. También hay en curso algunas conversaciones con la FDA en relación al estudio propuesto de Sarepta para evaluar un régimen adicional de inmunosupresión hacia la re-inclusión no ambulatoria en la etiqueta.
- Progreso de la línea de productos para varios programas de ARNpi: Se espera realizar la lectura de los estudios de Fase 1/2 para la distrofia muscular facioescapulohumeral (FSHD, por sus siglas en inglés)y la distrofia miotónica tipo 1, para ambas cohortes de dosis única ascendiente (SAD, por sus iniciales en inglés) y la dosis múltiple ascendente (MAD, por sus iniciales en inglés) que se esperan a inicios de 2026.
- Distrofia muscular facioescapulohumeral (FSHD): la inscripción de la SAD en el estudio de Fase 1/2 de la SRP-1001 ha finalizado y la cohorte 6 de la MAD está en curso.
- Distrofia miotónica tipo 1 (DM1): la inscripción de la SAD en el estudio de Fase 1/2 de la SRP-1003 ha finalizado y la cohorte 4 de la MAD está en curso.
- Enfermedad de Huntington (HD, por sus iniciales en inglés): en camino a iniciar el ensayo clínico para la SRP-1005 a finales de 2025 utilizando una ruta subcutánea de administración.
- Selección de objetivos de la investigación: tres nuevos objetivos han sido seleccionados además del candidato con DM1 de segunda generación que se seleccionó al cierre del tratado con Arrowhead Pharmaceuticals.
- World Muscle Society (WMS, por sus siglas en inglés), edición 2025 : En la reunión de la WMS, Sarepta presentó los nuevos datos de ELEVIDYS, así como las actualizaciones de nuestros programas LGMD 2E y PMO. Además, se presentaron varios estudios independientes, incluyendo los análisis preliminares de seguridad, tolerabilidad, y eficacia de un protocolo profiláctico de sirolimus para pacientes recibiendo terapia génica con delandistrogene moxeparvovec-rokl. Todos los pósters y presentaciones de la renunión de la WMS están disponibles en el sitio web de la compañía haciendo click aquí.
- Base financiera: la compañía ha tomado algunos pasos este trimestre para fortalecer su base financiera al extender la madurez de una parte significativa de sus notas convertibles a 2030, mejorando la liquidez con la disposición de su inversión de capital de Arrowhead Pharmaceuticals, y alcanzando ahorros de gastos por sobre los objetivos de reestructuración de costes declarados.
Información de la conferencia por teléfono
El evento será transmitido en vivo en la sección de relaciones con inversores del sitio web de Sarepta en el siguiente enlace: https://investorrelations.sarepta.com/events-presentations y luego del evento se archivará una reproducción por un año. Puede acceder a este evento haciendo click aquí.
Aspectos financieros destacados del tercer trimestre del 2025
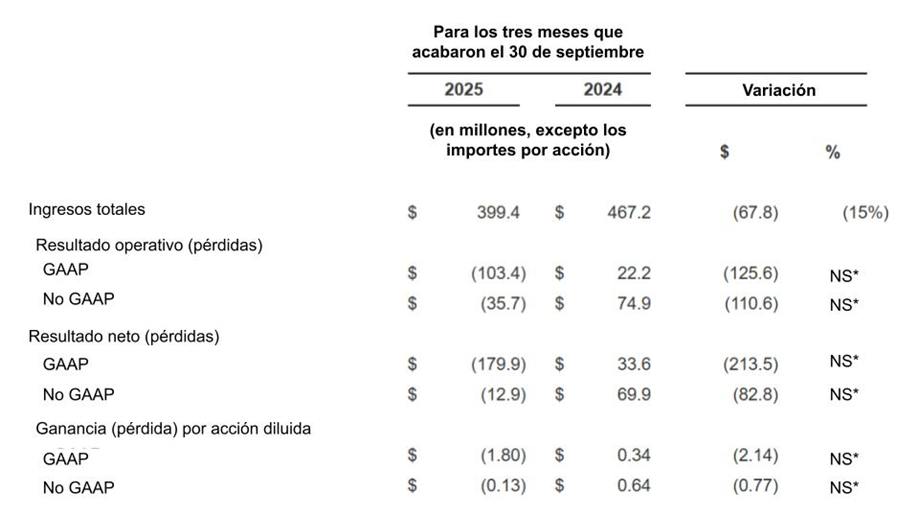
*NS: no significativo[1] Para obtener una explicación del uso de nuestras medidas económicas no GAAP, por favor revise la sección “Uso de Medidas Financieras no GAAP” que se encuentra más abajo en este comunicado. También, para ver la conciliación entre cada medida financiera GAAP y su correspondiente medida no GAAP más comparable, consulte las tablas al final de este comunicado.
| Efectivo, equivalentes de efectivo, efectivo restringido e inversiones a corto y largo plazo. |  |
Ingresos
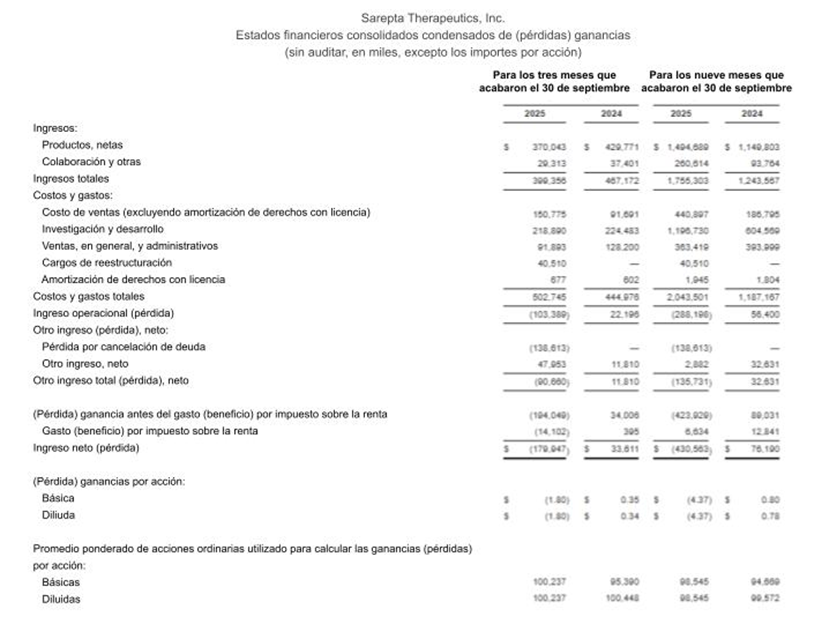
Los ingresos totales fueron de $399.4 millones por los tres meses que finalizaron el 30 de septiembre de 2025, en comparación con los $467.2 millones por el mismo periodo de 2024, una disminución de $67.8 millones. La disminución principalmente refleja $49.5 millones menos en los ingresos netos por producto de ELEVIDYS como un resultado de un volumen más vajo tras nuestra decisión de suspender los envíos de ELEVIDYS para pacientes no ambulatorios en EE. UU. en junio de 2025. Además, otros ingresos disminuyeron $8.1 millones principalmente debido a los ingresos por contrato de manufacturación que disminuyeron $9.2 millones por el volumen más bajo de envíos de ELEVIDYS a Roche.
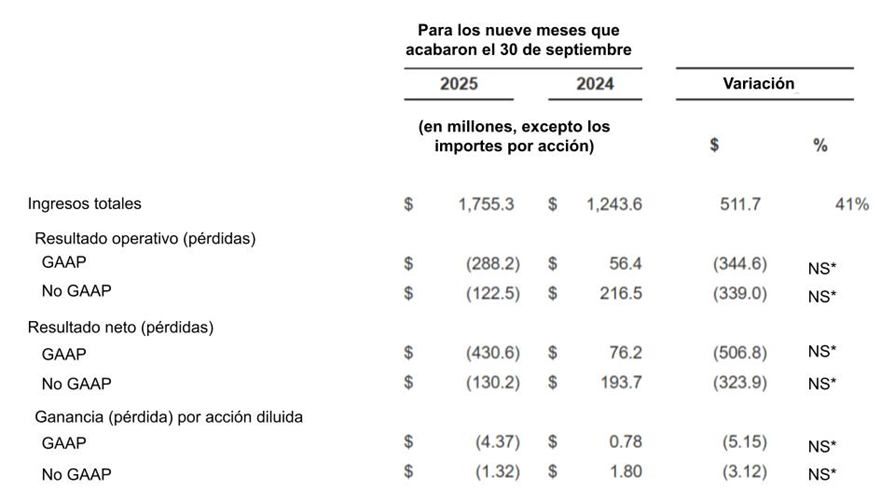
Los ingresos totales fueron $1,755.3 millones por los nueve meses que finalizaron el 30 de septiembre de 1015, en comparación con los $1,243.6 millones por el mismo periodo de 2024, un aumento de $511.7 millones. El incremento refleja principalmente $351.7 millones más en ingresos netos por producto de ELEVIDYS como resultado de su aprobación de etiqueta extendida en junio de 2024. Además, la colaboración y otros ingresos aumentaron aproximadamente $166.9 millones principalmente en relación a los $63.5 millones de ingresos de colaboración reconocidos como resultado de una aprobación regulatoria en Japón y los $112.0 millones de ingresos de colaboración reconocidos en relación al vencimiento de una opción por un programa de Roche durante los nueve meses que finalizaron el 30 de septiembre de 2025, en comparación con los $48.0 millones de ingresos por colaboración reconocidos en el mismo periodo de 2024 en relación a la opción rechazada de adquirir ciertos derechos ex-EE.UU. de un programa de desarrollo para Duchenne en etapa temprana y externo. Igualmente, los ingresos por contrato de manufacturación e ingresos por regalías aumentaron $19.4 millones y $9.9 millones, respectivamente, asociados con un aumento en la distribución de ELEVIDYS a Roche, así como ingresos por regalías de las ventas de ELEVIDYS por Roche, respectivamente.
Costo de ventas (excluyendo amortización de derechos licenciados)
Los costos de ventas (excluyendo amortización de derechos licenciados)fueron $150.8 millones por los tres meses que finalizaron el 30 de septiembre de 2025, en comparación con los $91.7 millones por el mismo periodo de 2024, un aumento de aproximadamente $59.1 millones. El aumento refleja principalmente la reducción del inventario de ELEVIDYS contabilizado anteriormente como gasto, el deterioro de los depósitos anticipados por fabricación y un aumento de las amortizaciones de determinados lotes de nuestros productos que no cumplieron nuestras especificaciones de calidad por los tres meses que finalizaron el 30 de septiembre de 2025, en comparación con el mismo periodo en 2024. El costo por ventas (excluyendo la amortización por derechos de licencia) fueron $440.9 millones por los nueve meses que finalizaron el 30 de septiembre de 2025, en comparación con los $186.8 millones por el mismo periodo en 2024, un aumento de $254.1 millones. El aumento principalmente refleja un incremento en la demanda de ELEVIDYS tras la aprobación de etiqueta extendida en junio de 2024, el deterioro del inventario de ELEVIDYS contabilizado anteriormente como gasto, un aumento en las amortizaciones de determinados lotes de nuestros productos que no cumplieron nuestras especificaciones de calidad, un aumento en los productos vendidos a Roche bajo el acuerdo de colaboración con Roche y el deterioro de los depósitos anticipados por fabricación por los nueve meses que finalizaron el 30 de septiembre de 2025, en comparación con el mismo periodo en 2024.
Gastos operativos y otros
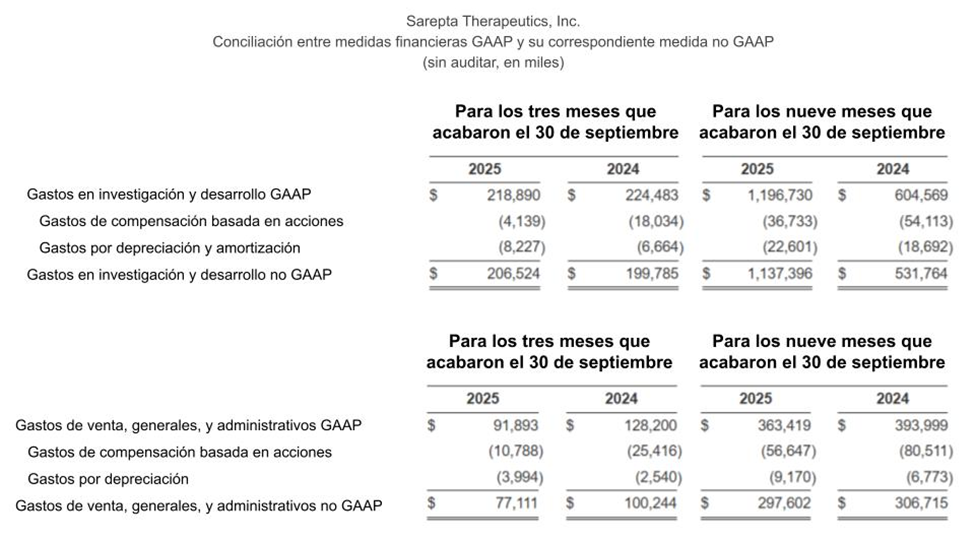
Los gastos en investigación y desarrollo fueron de $218.9 millones por los tres meses que finalizaron el 30 de septiembre de 2025, en comparación con los $224.5 millones por el mismo periodo en 2024, una disminución de $5.6 millones. La disminución se debe principalmente a la disminución de $102.7 millones en los gastos de fabricación predominantemente como resultado de los costos asociados con la terminación de nuestro acuerdo de desarrollo, fabricación comercial y suministro con Brammer Bio MA, LLC, un afiliado de la compañia Thermo Fisher Scientific, Inc. (Acuerdo Thermo) durante los tres meses que finalizaron el 30 de septiembre de 2024, sin actividad similar por los tres meses que finalizaron el 30 de septiembre de 2025, así como una disminución en compensación, otro personal, y gastos de compensación basada en acciones, como resultado de nuestro plan de reestructuración anunciado durante los tres meses que finalizaron el 30 de septiembre de 2025. Esta disminución fue parcialmente compensada por una disminución de $100.0 millones en gastos iniciales e intermedios debido a nuestro pago intermedio a Arrowhead, motivado por el logro de Arrowhead del primero de dos objetivos de reclutamiento predeterminados y la subsecuente autorización de la dosis escalada en un estudio de Fase 1/2 para el programa DM1, sin actividad similar por los tres meses que finalizaron el 30 de septiembre de 2024. Para los tres meses que finalizaron el 30 de septiembre de 2025, los gastos por investigación No GAAP y desarrollo fueron de $206.5 millones, en comparación con $199.8 millones por el mismo periodo de 2024, un aumento de $6.7 millones.
Los gastos por investigación y desarrollo fueron de $1,196.7 millones por los nueve meses que finalizaron el 30 de septiembre de 2025, en comparación con los $604.6 millones por el mismo periodo de 2024, un aumento de aproximadamente $592.1 millones. El aumento refleja principalmente un aumento de $538.6 millones en gastos iniciales e intermedios asociados con el acuerdo de licencia y colaboración, y el acuerdo de compraventa de acciones con Arrowhead, así como el pago intermedio de $100.0 millones hecho a Arrowhead por los nueve meses que finalizaron el 30 de septiembre de 2025, compensado parcialmente por una disminución de 74,9 millones de dólares en los gastos de fabricación principalmente debido a la terminación del Acuerdo Thermo durante los nueve meses que finalizaron el 30 de septiembre de 2024, así como a una disminución en la compensación, otro personal, y gastos de compensación, y gastos de compensación basada en acciones como resultado de nuestro plan de reestructuración en julio de 2025. Por los nueve meses que finalizaron el 30 de septiembre de 2025, los gastos por investigación no GAAP y desarrollo fueron de $1,137.4 millones, en comparación con los $531.8 millones por el mismo período de 2024, un aumento de $605.6 millones.
Los gastos por ventas, generales, y administrativos fueron de $91.9 millones para los tres meses que finalizaron el 30 de septiembre de 2025, en comparación con los $128.2 millones por el mismo período en 2024, una disminución de $36.3 millones. La disminución es principalmente impulsada por un compensación menor y otros gastos en personal debido a la reducción del número de empleados, así como a una disminución en la compensación basada en acciones debido a la reversión de gasto previamente reconocido relacionado con complementos salariales no consolidables, así como un resultado de nuestro plan de reestructuración anunciado en julio de 2025. Para los tres meses finalizados el 30 de septiembre de 2025, los gastos por ventas no GAAP, generales y administrativos fueron de $77.1 millones, en comparación con los $100.2 millones por el mismo período en 2024, una disminución de $23.1 millones.
Los gastos por ventas, generales, y administrativos fueron de $363.4 millones por los nueve meses que finalizaron el 30 de septiembre de 2025, en comparación con los $394.0 millones por el mismo período en 2024, una disminución de $30.6 millones. La disminución es principalmente impulsada por la reducción en el número de empleados como resultado de nuestro plan de reestructuración por los nueve meses que finalizaron el 30 de septiembre de 2025 y una disminución neta en la compensación basada en acciones principalmente debido al logro de las condiciones de desempeño en relación a determinadas Unidades de Acciones por Desempeño (PSUs, por sus iniciales en inglés) durante los nueve meses que finalizaron el 30 de septiembre de 2024, y la reversión del gasto previamente reconocido relacionado con complementos salariales no consolidables como resultado de nuestro plan de reestructuración anunciado en julio de 2025, compensado parcialmente por el cumplimiento de las condiciones de servicio restantes asociadas con ciertas PSUs en marzo de 2025. Para los nueve meses que finalizaron el 30 de septiembre de 2025, los gastos por ventas no GAAP, generales y administrativos fueron de $297.6 millones, en comparación con los $306.7 millones por el mismo periodo en 2024, una disminución de $9.1 millones.
Los cargos por reestructuración fueron de $40.5 millones por los tres y nueve meses que finalizaron el 30 de septiembre de 2025, sin actividad similar por los mismos períodos en 2024. Estos cargos estuvieron relacionados principalmente con beneficios por despido de empleados, incluyendo indemnizaciones, así como con la depreciación acelerada de activos afectados por nuestro plan de reestructuración, anunciado en julio de 2025, cuyo objetivo fue reducir los gastos operativos y alinear nuestra estructura de costos con las prioridades estratégicas, con el fin de mejorar la flexibilidad financiera y cumplir con nuestras obligaciones financieras para 2027.
La pérdida por extinción de deuda fue de $138.6 millones por los tres y nueve meses que finalizaron el 30 de septiembre de 2025, sin actividad similar por los mismos periodos en 2024. La pérdida por extinción de deuda es un resultado de nuestro refinanciamiento parcial de los bonos con vencimiento en 2027 a través de una transacción de intercambio negociada de forma privada donde se intercambiaron $700.0 millones en monto principal agregado de dichos bonos por: (1) $602.0 millones en monto principal agregado de nuevos bonos convertibles sénior con vencimiento el 1 de septiembre de 2030 (los “Bonos 2030”), netos de costos de emisión por $13.4 millones; (2) pagos en efectivo por $127.3 millones, incluidos $4.0 millones en intereses devengados; y (3) la emisión de $5.9 millones de acciones ordinarias de la compañía con un valor razonable de mercado aproximado de $107.3 millones, neto de costos de emisión por $2.4 millones.
Otros ingresos, netos por los tres meses que finalizaron el 30 de septiembre de 2025 y 2024 fue de $48.0 millones y $11.8 millones, respectivamente. El cambio refleja principalmente los ajustes del valor razonable de nuestros inversores, incluyendo Arrowhead, durante los tres meses que finalizaron el 30 de septiembre de 2025. Otros ingresos, los netos por los nueve meses que finalizaron el 30 de septiembre de 2025 y 2024, fueron de $2.9 millones y $32.6 millones, respectivamente. El cambio refleja principalmente la disminución en ingresos por intereses debido a cambios en la combinación de inversiones de nuestro portafolio de inversiones y los ajustes del valor razonable de nuestras inversiones con Arrowhead durante los nueve meses que finalizaron el 30 de septiembre de 2025.
Gastos (beneficio) por impuesto sobre la renta por los tres meses que finalizaron el 30 de septiembre de 2025 y 2024, fue de $(14.1) millones y $0.4 millones, respectivamente. El gasto por impuesto sobre la renta por los nueve meses que finalizaron el 30 de septiembre de 2024 y 2024, fue de $6.6 millones y $12.8 millones, respectivamente. El gasto por ingreso sobre la renta beneficio por los tres meses que finalizaron el 30 de septiembre de 2025, está relacionado con el impuesto sobre la renta registrado en el trimestre a la fecha la cual compensa el gasto por impuestos registrado en la rentabilidad en períodos interinos previos. El gasto por impuesto sobre la renta por los nueve meses que finalizaron el 30 de septiembre de 2025, así como por los tres y nueve meses que finalizaron el 30 de septiembre de 2024, está relacionado principalmente a los impuestos sobre la renta estatales, federales y extranjeros por los cuales las pérdidas fiscales o los créditos fiscales disponibles no lo estaban para compensar.
Uso de medidas no GAAP
Además de las medidas económicas GAAP expuestas en este comunicado de prensa, hemos incluído las siguientes medidas no GAAP:
- Nosotros definimos las ganancias (pérdidas) netas no GAAP como ganancias (pérdidas) netas GAAP, excluyendo ganancias/gastos netos por depreciación y amortización, y gasto por compensación basado en acciones, cargos por reestructuración, otros aspectos y el impacto estimado en el impuesto sobre la renta de cada ajuste no GAAP antes de impuestos.
- Nosotros definimos las pérdidas netas no GAAP por acción como pérdidas netas no GAAP, como se definieron antes, divididas por el promedio ponderado de acciones ordinarias en circulación, dado que la inclusión de equivalentes dilutivos de acciones ordinarias en circulación resulta anti-dilutiva. Nosotros definimos las ganancias no GAAP por acción como ingreso neto no GAAP, como se definió antes, dividido por el promedio ponderado de acciones ordinarias en circulación, ajustado por la inclusión de acciones adicionales bajo el método “si se convirtiera”, si fuera aplicable y no resultara anti-dilutivo.
- Nosotros definimos las ganancias (pérdidas) por operaciones no GAAP como ganancias (pérdidas) por operaciones GAAP excluyendo el gasto por depreciación y amortización, gasto por compensación basado en acciones, y cargos por reestructuración.
- Nosotros definimos los gastos por investigación y desarrollo no GAAP como gastos por investigación y desarrollo GAAP, excluyendo el gasto por depreciación y amortización, y el gasto por compensación basado en acciones.
- Nosotros definimos los gastos de venta, generales, y administrativos no GAAP como gastos de venta, generales, y administrativos GAAP, excluyendo gasto por depreciación, gasto por compensación basada en acciones, y otros gastos.
Los siguientes componentes se usaron para ajustar nuestras medidas financieras GAAP en las medidas no GAAP previamente definidas:
- Interés, depreciación y amortización: los ingresos (gastos) por intereses, cantidades netas pueden variar sustancialmente de periodo a periodo debido a cambios en el efectivo y balances de deuda, y ramos de interés impulsados por las condiciones en el mercado fuera de nuestras operaciones. El gasto por depreciación puede variar sustancialmente de periodo a periodo pues las compras de propiedad y equipo pueden variar significativamente de periodo a periodo sin ninguna correlación directa a nuestro desempeño de operaciones. El gasto por amortización asociado principalmente con costos de patente son amortizados en un periodo de varios años después de la adquisición, aplicación de la patente, o renovación.
- Gastos por compensación basada en acciones: los gastos por compensación basada en acciones representan cargos de no efectivo relacionados con premios en acciones que hemos entregado. Aunque estos son cargos recurrentes a las operaciones, creemos que la medida de estas cantidades puede variar sustancialmente de periodo a periodo y depende significativamente de factores que no son una consecuencia directa del desempeño por operaciones que esté en nuestro control. Por ello, creemos que excluir estos cargos facilita la comparación de nuestro desempeño operacional en periodos diferentes.
- Los cargos por reestructuración no tienen una correlación directa con las operaciones de negocios futuras, ni tampoco los cargos registrados resultantes reflejan el desempeño de nuestras operaciones en curso para el período en el cual tales cargos fueron registrados. Además, los cargos por reestructuración no son considerados como gastos por operación normales debido a la variabilidad de cantidades y la falta de predictibilidad en cuanto a su ocurrencia y/o momento.
- Otros elementos: evaluamos otros elementos de gasto e ingreso de manera individual. Tomamos en cuenta características cuantitativas y cualitativas de cada elemento, incluyendo la naturaleza, si los elementos se relacionan con nuestras operaciones de negocios en curso, y si esperamos que los elementos continúen o ocurran de manera regular. Estos otros elementos incluyen la ganancia/pérdida de inversiones estratégicas, cambios en el valor razonable de los derivados, cargos por reestructuración, y pérdida por cancelación de deuda, y podrán incluir otros que encajen con las características previas en el futuro. Excluimos de nuestros resultados no GAAP lo siguiente:
- La (ganancia) pérdida en inversiones estratégicas como resultado de tales ganancias y pérdidas no son representativas de nuestras operaciones de negocios normales, las cuales según, harían que fuera difícil comparar nuestros resultados con compañías del mismo sector que también dan información no sujeta a normas GAAP (no GAAP). Estamos realizando este cambio empezando en 2025 porque, como nuestras inversiones estratégicas han aumentado, reconocemos que la variabilidad resultante puede impedir la comparabilidad entre periodos de nuestro desempeño financiero para nuestras operaciones de negocios en curso.
- La variación en el valor razonable de los derivados relacionados con pagos contingentes sujetos a regulación (que cumplen con la definición de derivado) a los accionistas vendedores de Myonexus Therapeutics, Inc., así como a una institución académica bajo un acuerdo de licencia independiente, se considera una partida no monetaria y no se clasifica como gasto operativo normal, debido a la variabilidad de las cantidades y a la falta de previsibilidad en cuanto a su ocurrencia y/o momento.
- La pérdida por extinción de deuda se considera un evento poco frecuente, ya que está asociada a una decisión financiera distinta y no indica el desempeño de nuestras operaciones centrales, lo cual dificulta la comparación de nuestros resultados con los de compañías del mismo sector que también dan información no sujeta a normas GAAP (no GAAP).
Utilizamos estas medidas no GAAP como indicadores clave de desempeño con el propósito de evaluar internamente el rendimiento operativo y los requerimientos de liquidez. Asimismo, consideramos que estas medidas no GAAP aumentan la comparabilidad de los resultados entre período y período, y son útiles para los inversores, pues proporcionan una base similar a la utilizada por la administración para evaluar nuestro desempeño. Estas medidas no GAAP no deben considerarse de forma aislada ni como reemplazo de la presentación de nuestros resultados financieros según las normas GAAP. El uso de términos como gastos de investigación y desarrollo no GAAP, gastos de venta, generales y administrativos no GAAP, resultado (pérdida) operativo no GAAP, ingreso (pérdida) neto no GAAP, y utilidad (pérdida) diluida por acción no GAAP pueden diferir de las medidas similares reportadas por otras compañías, lo que podría limitar la comparabilidad, y no están basadas en un conjunto integral de reglas o principios contables. Todas las medidas no GAAP relevantes se concilian con sus respectivas medidas GAAP en la tabla adjunta “Conciliación de medidas financieras GAAP con medidas financieras no GAAP”.
Sobre EXONDYS 51
EXONDYS 51 usa tecnología química y de omisión de exón propiedad de Sarepta denominada morfolino fosforodiamidato (PMOs, por sus iniciales en inglés) para unirse al exón 51 de distrofina pre-ARNm, resultando en la exclusión, u “omisión”, de este exón durante el procesamiento del ARNm en pacientes con mutaciones genéticas que son susceptibles a la omisión del exón 51. La omisión de exón está pensada para permitir la producción de una proteína distrofina truncada de manera interna.
EXONDYS 51 está indicada para el tratamiento de la distrofia muscular de Duchenne (DMD) en pacientes que tienen una mutación confirmada del gen DMD que es susceptible a la omisión del exón 51. Esta indicación está aprobada bajo aprobación acelerada basada en un aumento de distrofina en el músculo esquelético observada en algunos pacientes tratados con EXONDYS 51. La aprobación continua de esta indicación podría ser supeditada a la verificación de un beneficio clínico en ensayos confirmatorios.
Información de seguridad importante sobre EXONDYS 51
Han habido reacciones de hipersensibilidad, incluyendo broncoespasmos, dolor en el pecho, tos, taquicardia, y urticaria, en pacientes quienes fueron tratados con EXONDYS 51. Si se presenta una reacción de hipersensibilidad, dé el tratamiento médico apropiado y considere retrasar la infusión o interrumpir la terapia con EXONDYS 51.
Las reacciones adversas en pacientes con DMD (N=8) tratados con EXONDYS 51 de 30 mg. o 50 mg/kg por semana por infusión intravenosa (IV) con una incidencia de al menos 25% más que el placebo (N=4) (Estudio 1, 24 semanas) fueron (EXONDYS 51, placebo): trastorno del equilibrio (38 %, 0 %), vómitos (38 %, 0 %) y dermatitis de contacto (25 %, 0 %). Las reacciones adversas más comunes fueron trastorno del equilibrio y vómitos. Debido al número pequeño de pacientes, éstas representan frecuencias brutas que podrían no reflejar las frecuencias observadas en la práctica. No se recomienda el régimen de dosis semanal de 50 mg/kg de EXONDYS 51.
Las reacciones adversas más comunes de los estudios clínicos observacionales (N=163) vistos en más del 10% de los pacientes fueron dolor de cabeza, tos, sarpullido, y vómito.
Podrían presentarse otros eventos adversos.
Para reportar SOSPECHAS DE REACCIONES ADVERSAS contacte a Sarepta Therapeutics, Inc. al 1-888-SAREPTA (1-888-727-3782) o la FDA al 1-800-FDA-1088 o visite www.fda.gov/medwatch.
Para más información, por favor revise la Información de receta en EE. UU. completa de EXONDYS 51 (eteplirsen).
Sobre VYONDYS 53
VYONDYS 53 (golodirsen) usa tecnología química y de omisión de exón propiedad de Sarepta denominada morfolino fosforodiamidato (PMOs, por sus iniciales en inglés) para unirse al exón 53 de distrofina pre-ARNm, resultando en la exclusión, u “omisión”, de este exón durante el procesamiento del ARNm en pacientes con mutaciones genéticas que son susceptibles a la omisión del exón 53. La omisión de exón está pensada para permitir la producción de una proteína distrofina truncada de manera interna.
VYONDYS 53 está indicada para el tratamiento de la distrofia muscular de Duchenne (DMD) en pacientes que tienen una mutación confirmada del gen DMD que es susceptible a la omisión del exón 53. Esta indicación está aprobada bajo aprobación acelerada basada en un aumento de distrofina en el músculo esquelético observada en algunos pacientes tratados con VYONDYS 53. La aprobación continua de esta indicación podría ser supeditada a la verificación de un beneficio clínico en ensayos confirmatorios.
VYONDYS 53 ha cumplido con los estándares estatutarios completos para la seguridad y efectividad y, por ello, no debe considerarse en investigación o experimental.
Información de seguridad importante sobre VYONDYS 53
CONTRAINDICACIONES: VYONDYS 53 está contraindicada en pacientes con una seria reacción de hipersensibilidad a golodirsen o a cualquiera de los ingredientes inactivos en VYONDYS 53. Se ha presentado anafilaxia en pacientes que recibieron VYONDYS 53.
ADVERTENCIAS Y PRECAUCIONES
Reacciones de hipersensibilidad: Se han presentado reacciones de hipersensibilidad, incluyendo anafilaxia, sarpullido, pirexia, prurito, urticaria, dermatitis y exfoliación cutánea en pacientes tratados con VYONDYS 53, algunos de ellos requiriendo de tratamiento. Si se presenta una reacción de hipersensibilidad, dé el tratamiento médico apropiado y considere retrasar la infusión, interrumpir o descontinuar la terapia con VYONDYS 53, y monitoree hasta que la condición se resuelva. VYONDYS 53 está contraindicada en pacientes con una seria reacción de hipersensibilidad a golodirsen o a cualquiera de los ingredientes inactivos en VYONDYS 53.
Toxicidad renal: se observó toxicidad renal en animales que recibieron golodirsen. Aunque la toxicidad renal no se observó en los estudios clínicos con VYONDYS 53, la experiencia clínica con VYONDYS 53 es limitada, y se ha observado toxicidad renal, incluyendo posible glomerulonefritis mortal, tras la administración de algunos oligonucleótidos antisentido. La función renal debe ser monitoreada en pacientes que toman VYONDYS 53. Debido al efecto de la masa músculo esquelética reducida en las medidas de creatinina, ésta podría no ser una medida confiable de la función renal en pacientes con DMD. Antes de iniciar el tratamiento con VYONDYS 53, se deben medir la cistatina C sérica, un análisis de orina con tira reactiva y la relación proteína/creatinina en orina. Considere también evaluar la tasa de filtración glomerular mediante un marcador exógeno de filtración antes de empezar a recibir VYONDYS 53.Durante el tratamiento, se debe monitorear el análisis de orina con tira reactiva cada mes, y la cistatina C sérica junto con la relación proteína/creatinina en orina cada tres meses. Para el monitoreo de proteínas en orina, sólo debe utilizarse orina que se espere esté libre de VYONDYS 53 excretado. Puede usarse orina recolectada el mismo día de la infusión con VYONDYS 53, antes de administrarse, o al menos 48 horas después de la última infusión. Alternativamente, puede utilizarse una prueba de laboratorio que no emplee el reactivo rojo de pirogalol, ya que este reactivo tiene el potencial de reaccionar de forma cruzada con VYONDYS 53 excretado en la orina y conducir así a un resultado falso positivo en la detección de proteínas en la orina.
Si se detecta un aumento persistente de la cistatina C sérica o la proteinuria, consulte a un nefrólogo pediátrico para una mayor evaluación.
REACCIONES ADVERSAS: Las reacciones adversas observadas en al menos el 20% de los pacientes tratados y más del placebo fueron (VYONDYS 53, placebo): dolor de cabeza (41%, 10%), pirexia (41%, 14%), caídas (29%, 19%), dolor abdominal (27%, 10%), nasofaringitis (27%, 14%), tos (27%, 19%), vómito (27%, 19%), y náuseas (20%, 10%).
Otras reacciones adversas que se presentaron con una frecuencia mayor del 5% de los pacientes tratados con VYONDYS 53 y que el placebo fueron: dolor en el lugar de la administración, dolor de espalda, dolor, diarrea, mareos, esguinces de ligamentos, contusión, influenza, dolor orofaríngeo, rinitis, abrasión cutánea, infección en el oído, alergia estacional, taquicardia, reacción en el lugar de colocación de catéter, estreñimiento, y fractura.
Podrían presentarse otros eventos adversos.
Para reportar SOSPECHAS DE REACCIONES ADVERSAS contacte a Sarepta Therapeutics, Inc. al 1-888-SAREPTA (1-888-727-3782) o la FDA al 1-800-FDA-1088 o visite www.fda.gov/medwatch.
Para más información, por favor revise la Información de receta en EE. UU. completa de VYONDYS 53 (golodirsen).
Sobre AMONDYS 45
AMONDYS 45 (casimersen) usa tecnología química y de omisión de exón propiedad de Sarepta denominada morfolino fosforodiamidato (PMOs, por sus iniciales en inglés) para unirse al exón 45 de distrofina pre-ARNm, resultando en la exclusión, u “omisión”, de este exón durante el procesamiento del ARNm en pacientes con mutaciones genéticas que son susceptibles a la omisión del exón 45. La omisión de exón está pensada para permitir la producción de una proteína distrofina truncada de manera interna.
AMONDYS 45 está indicada para el tratamiento de la distrofia muscular de Duchenne (DMD) en pacientes que tienen una mutación confirmada del gen DMD que es susceptible a la omisión del exón 45. Esta indicación está aprobada bajo aprobación acelerada basada en un aumento de distrofina en el músculo esquelético observada en algunos pacientes tratados con AMONDYS 45. La aprobación continua de esta indicación podría ser supeditada a la verificación de un beneficio clínico en ensayos confirmatorios.
AMONDYS 45 ha cumplido con los estándares estatutarios completos para la seguridad y efectividad y, por ello, no debe considerarse en investigación o experimental.
Información de seguridad importante sobre AMONDYS 45
CONTRAINDICACIONES: AMONDYS 45 está contraindicada en pacientes con una seria reacción de hipersensibilidad a casimersen o a cualquiera de los ingredientes inactivos en AMONDYS 45. Se han presentado momentos de hipersensibilidad incluyendo angioedema y anafilaxia.
ADVERTENCIAS Y PRECAUCIONES
Hipersensibilidad: Se han presentado reacciones de hipersensibilidad, incluyendo anafilaxia y angioedema, en pacientes tratados con AMONDYS 45. Si se presenta una reacción de hipersensibilidad, dé el tratamiento médico apropiado y considere retrasar la infusión, interrumpir o descontinuar la infusión con AMONDYS 45, y monitoree hasta que la condición se resuelva. AMONDYS 45 está contraindicada en pacientes con una seria reacción de hipersensibilidad a casimersen o a cualquiera de los ingredientes inactivos en AMONDYS 45.
Toxicidad renal: se observó toxicidad renal en animales que recibieron casimersen. Aunque la toxicidad renal no se observó en los estudios clínicos con AMONDYS 45, se ha observado toxicidad renal, incluyendo posible glomerulonefritis mortal, tras la administración de algunos oligonucleótidos antisentido. La función renal debe ser monitoreada en pacientes que toman AMONDYS 45. Debido al efecto de la masa músculo esquelética reducida en las medidas de creatinina, ésta podría no ser una medida confiable de la función renal en pacientes con DMD. Antes de iniciar el tratamiento con AMONDYS 45, se deben medir la cistatina C sérica, un análisis de orina con tira reactiva y la relación proteína/creatinina en orina. Considere también evaluar la tasa de filtración glomerular mediante un marcador exógeno de filtración antes de empezar a recibir AMONDYS 45. Durante el tratamiento, se debe monitorear el análisis de orina con tira reactiva cada mes, y la cistatina C sérica junto con la relación proteína/creatinina en orina cada tres meses. Para el monitoreo de proteínas en orina, sólo debe utilizarse orina que se espere esté libre de AMONDYS 45 excretado. Puede usarse orina recolectada el mismo día de la infusión con AMONDYS 45, antes de administrarse, o al menos 48 horas después de la última infusión. Alternativamente, puede utilizarse una prueba de laboratorio que no emplee el reactivo rojo de pirogalol, ya que este reactivo tiene el potencial de reaccionar de forma cruzada con AMONDYS 45 excretado en la orina y conducir así a un resultado falso positivo en la detección de proteínas en la orina.
Si se detecta un aumento persistente de la cistatina C sérica o la proteinuria, consulte a un nefrólogo pediátrico para una mayor evaluación.
REACCIONES ADVERSAS: Las reacciones adversas observadas en al menos el 20% de los pacientes tratados con AMONDYS 45 y al menos 5% más frecuentemente que el grupo del placebo fueron (AMONDYS 45, placebo): infecciones de las vías respiratorias superiores (65 %, 55 %), tos (33 %, 26 %), pirexia (33 %, 23 %), dolor de cabeza (32 %, 19 %), artralgia (21 %, 10 %) y dolor orofaríngeo (21 %, 7 %).
Otras reacciones adversas que se presentaron en al menos 10% de los pacientes tratados con AMONDYS 45 y al menos 5% más frecuentemente que en el grupo del placebo fueron: dolor de oído, náuseas, infección en el oído, dolor post-traumático, y mareos y aturdidos.
Podrían presentarse otros eventos adversos.
Para reportar SOSPECHAS DE REACCIONES ADVERSAS contacte a Sarepta Therapeutics, Inc. al 1-888-SAREPTA (1-888-727-3782) o la FDA al 1-800-FDA-1088 o visite www.fda.gov/medwatch.
Para más información, por favor revise la Información de receta en EE. UU. completa de VYONDYS 53 (golodirsen).
Sobre ELEVIDYS (delandistrogene moxeparvovec-rokl)
ELEVIDYS (delandistrogene moxeparvovec-rokl) es una terapia de transferencia de genes basada en virus adeno-asociado (AAV) de una sola dosis para infusión intravenosa diseñada para abordar la causa genética subyacente de la distrofia muscular de Duchenne (mutaciones o cambios en el gen DMD que resultan en la falta de la proteína distrofina) a través de la entrega de un transgén que codifica para la producción objetivo de la micro distrofina ELEVIDYS en el músculo esquelético.
ELEVIDYS está indicada para el tratamiento de la distrofia muscular de Duchenne (DMD) en personas de al menos 4 años de edad.
- Para pacientes ambulatorios y que tienen una mutación confirmada en el gen DMD.
- Para pacientes no ambulatorios y que tienen una mutación confirmada en el gen DMD.
La indicación DMD en pacientes no ambulatorios está aprobada bajo aprobación acelerada basada en la expresión de micro distrofina ELEVIDYS en el músculo esquelético. La aprobación continua de esta indicación puede ser eventual hasta la verificación y descripción del beneficio clínico en un ensayo (o ensayos) confirmatorio.
Información importante de seguridad
CONTRAINDICACIÓN: ELEVIDYS está contraindicada en pacientes con cualquier eliminación en el exón 8 y/o exón 9 en el gen DMD.
ADVERTENCIAS Y PRECAUCIONES
Reacciones relacionadas con la infusión:
- Han habido reacciones relacionadas con la infusión, incluyendo reacciones de hipersensibilidad y anafilaxia, durante o hasta después de varias horas tras la administración de ELEVIDYS. Monitorear de cerca a los pacientes durante la administración y por al menos 3 horas después de acabar la infusión. Si los síntomas de reacciones relacionadas con la infusión se presentan, retrasar, o detener la infusión y dar el tratamiento adecuado. Una vez que los síntomas se resuelvan, la infusión puede volver a empezar a un ritmo más lento.
- Se debe administrar ELEVIDYS en un espacio donde el tratamiento para reacciones relacionadas con la infusión esté disponible inmediatamente.
- Descontinuar la infusión por anafilaxia.
Lesión hepática severa aguda:
- Se ha observado lesión hepática severa aguda con ELEVIDYS, y la administración puede resultar en enzimas hepáticas elevadas (como GGT, GLDH, ALT, AST) o bilirrubina total, normalmente vista dentro de las 8 semanas.
- Los pacientes con deterioro hepático, condición hepática crónica, o enfermedad hepática aguda (por ejemplo, infección viral hepática aguda) pueden estar en mayor riesgo de lesión hepática severa aguda. Posponer la administración de ELEVIDYS en pacientes con enfermedad hepática aguda hasta que se resuelva o se controle.
- Previo a la administración de ELEVIDYS, llevar a cabo pruebas de enzimas hepáticas y monitorear la función hepática (examen clínico, GGT, y bilirrubina total) semanalmente durante los primeros 3 meses luego de la infusión de ELEVIDYS. Continuar monitoreando si así se indica clínicamente, hasta que los resultados sean normales (examen clínico normal, GGT, y los niveles de bilirrubina total regresen cerca de los niveles de referencia).
- Se recomienda el tratamiento sistémico con corticosteroides para pacientes antes y después de la infusión de ELEVIDYS. Ajustar el régimen de corticosteroides cuando se indique. Si se sospecha de lesión hepática severa aguda, se recomienda consultarlo con un especialista.
Miositis inmunomediada:
- En ensayos clínicos, se ha observado miositis inmunomediada aproximadamente 1 mes después de la infusión de ELEVIDYS en pacientes con mutación por eliminación involucrando exón 8 y/o exón 9 en el gen DMD. Se observaron síntomas de debilidad muscular severa, incluyendo disfagia, disnea e hipofonía.
- Se encuentran disponibles datos limitados para el tratamiento con ELEVIDYS en pacientes con mutaciones en el gen DMD en exones 1 al 17 y/o exones 59 al 71. Los pacientes con eliminaciones en estas regiones pueden estar en riesgo de una reacción severa de miositis inmunomediada.
- Recomendar a los pacientes contactar a un médico inmediatamente si experimentan cualquier color muscular incrementado sin explicar, sensibilidad, o debilidad, incluyendo disfagia, disnea, o hipofonía, pues éstos podrían ser síntomas de miositis. Considerar tratamiento inmunomodulador adicional (inmunosupresores, por ejemplo un inhibidor de calcineurina, además de los corticosteroides) basado en la presentación clínica del paciente y el historial médico, si estos síntomas se presentan.
Miocarditis:
- Se han observado miocarditis aguda seria y elevaciones de troponina-I seguidas de infusión de ELEVIDYS en ensayos clínicos.
- Si un paciente experimenta miocarditis, aquellos con deterioro preexistente de la fracción de eyección del ventrículo izquierdo (FEVI) pueden estar en mayor riesgo de resultados adversos. Monitorear troponina-I antes de la infusión de ELEVIDYS y semanalmente durante el primer mes seguido de la infusión, y continuar monitoreando si así se indica clínicamente. El monitoreo frecuente puede asegurarse en presencia de síntomas cardíacos, como dolor en el pecho o dificultad para respirar.
- Recomendar a los pacientes contactar a un médico inmediatamente si experimentan síntomas cardiacos.
Inmunidad preexistente contra AAVrh74:
- En terapias génicas basadas en vector VAA, los anticuerpos anti VAA preexistentes pueden impedir la expresión transgén en los niveles terapéuticos deseados. Luego del tratamiento con ELEVIDYS, todos los pacientes desarrollaron anticuerpos anti AAVrh74.
- Llevar a cabo pruebas iniciales para detectar la presencia de anticuerpos totales de unión anti AAVrh74 previas a la administración de ELEVIDYS.
- No se recomienda la administración de ELEVIDYS en pacientes con valores elevados de anticuerpos totales de unión anti AAVrh74 a más o igual que 1:400.
Reacciones adversas:
- Las reacciones adversas más comunes (incidencia igual o superior a 5%) que se reportaron en estudios clínicos fueron vómitos, náuseas, lesión hepática, pirexia, y trombocitopenia.
Reporte con la FDA los efectos secundarios negativos a medicamentos recetados. Visite
www.fda.gov/medwatch o llame al 1-800-FDA-1088. Puede también reportar efectos secundarios con Sarepta Therapeutics al 1-888-SAREPTA (1-888-727-3782).
Para más información, por favor revise la Información de receta completa de ELEVIDYS (delandistrogene moxeparvovec-rokl).
Acerca de Sarepta Therapeutics
Sarepta está en una misión urgente: diseñar medicina genética de precisión para enfermedades raras que devastan vidas y acortan futuros. Somos líderes en la distrofia muscular de Duchenne (Duchenne) y estamos construyendo un portafolio amplio de programas sobre el sistema nervioso central y muscular, así como enfermedades cardiacas. Para más información, por favor visite:
www.sarepta.com o síganos en LinkedIn, X, Instagram, y Facebook.
Declaraciones a futuro
Para proporcionar a los inversores de Sarepta una comprensión de sus resultados actuales y futuros prospectos, este comunicado de prensa contiene declaraciones que son a futuro. Cualquier declaración en este comunicado de prensa que no sea de hecho histórico puede ser considerada como declaración a futuro. Palabras como “creer”, “anticipar”, “planear”, “esperar”, “será”, “puede”, “pretende”, “prepara”, “observa”, “potencial”, “posible” y expresiones similares tienen la intención de identificar declaraciones a futuro. Estas declaraciones a futuro incluyen declaraciones en relación a nuestras operaciones futuras, desempeño financiero y proyecciones, planes de negocios, oportunidades de comercialización, prioridades y programas de investigación y desarrollo; nuestros ensayos clínicos planeados y en curso; los planes e hitos esperados de nuestro ensayo confirmatorio ESSENCE y una opción para la aprobación tradicional de casimersen y golodirsen, así como nuestras expectativas en relación a la reducción etiqueta de ELEVIDYS.
Estas declaraciones a futuro involucran riesgos e incertidumbres, muchas de las cuales están más allá del control de Sarepta. Los resultados reales pudieran diferir materialmente de aquellos mencionados o dados a entender por estas declaraciones a futuro como resultado de tales riesgos e incertidumbres. Los factores de riesgo conocidos incluyen lo siguiente: podríamos no ser capaces de cumplir con todos los compromisos posteriores a la aprobación de la FDA y los requerimientos respecto a nuestros productos a tiempo o en absoluto; los datos principales se basan en el análisis preliminar y podrían diferir materialmente de los datos iniciales; podríamos no ser capaces de lograr una alineación con la FDA en relación a una opción para la aprobación tradicional para casimersen y golodirsen, incluyendo alguna limitación en la fiabilidad de la FDA en la evidencia del mundo real; la reducción del personal puede tomar más tiempo o resultar en más cargos importantes o expedición de fondos que lo anticipado, o de otro modo impactar negativamente a la Compañía y sus planes de negocios durante y después del periodo en el que se ejecute la reducción de personal; el éxito de los ensayos clínicos, especialmente si están basados en una muestra de pacientes pequeña, no aseguran que ensayos clínicos posteriores serán exitosos, y los resultados de investigación futura pudieran no ser consistentes con resultados positivos pasados, o pudieran no cumplir con los requisitos obligatorios reguladores para la seguridad y eficacia de los productos candidatos; ciertos programas podrían nunca alcanzar en el área clínica o podrían ser descontinuados por una variedad de razones, incluyendo que los reguladores impusieran una pausa clínica y que nosotros suspendiéramos o terminaramos la investigación clínica o los ensayos; si el número real de pacientes afectados por enfermedades al que apuntamos tratar es más pequeño de lo estimado, o nuestros ingresos y habilidad para lograr la rentabilidad podrían ser afectados de manera adversa; podríamos no ser capaces de ejecutar nuestros planes de negocios, incluyendo el cumplir con nuestros hitos regulatorios esperados o planeados y tiempos para ello, los planes de investigación y desarrollo clínico, y de llevar nuestros productos candidatos al mercado, por varias razones, algunas de las cuales pudieran estar fuera de nuestro control, incluyendo limitaciones de recursos económicos y otros por parte de la compañía, limitaciones de fabricación que podrían no ser anticipadas o resueltas a tiempo, y decisiones regulatorios, de la corte o alguna agencia, como las decisiones hechas por la Oficina de Patentes y Marcas Registradas de los Estados Unidos en relación a las patentes que cubren nuestros productos candidatos, el impacto del cierre del gobierno federal en la FDA; y aquellos riesgos identificados bajo el encabezado “Factores de riesgo” en nuestro Reporte Anual más reciente sobre la Forma 10-Q para el año finalizado el 31 de diciembre de 2024, y nuestro Reporte Trimestral más reciente sobre la Forma 10-Q archivada con la Comisión de Bolsa y Valores (SEC, por sus siglas en inglés) así como otros archivos SEC hechos por la Compañía, los cuales le invitamos a revisar.
Información publicada en Internet
De manera rutinaria publicamos información en internet que puede ser importante para inversores en la sección “Para Inversores” de nuestro sitio web www.sarepta.com. Invitamos a inversores y potenciales inversores a que consulten nuestro sitio web regularmente para conocer información importante sobre nosotros.
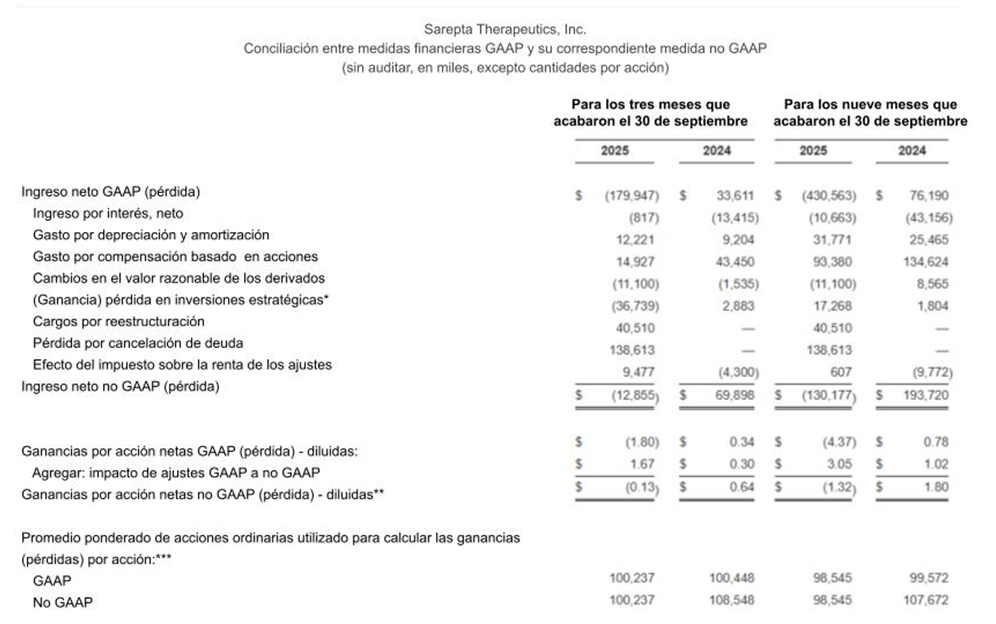
*Empezando en el primer trimestre de 2025, las pérdidas (ganancias) en inversiones estratégicas se incluyeron como una medida no GAAP para ajustar nuestras medidas financieras GAAP. Los resultados financieros no GAAP correspondientes a los tres y nueve meses que finalizaron el 30 de septiembre de 2024 se han actualizado para reflejar este cambio y facilitar la comparabilidad. Para más detalles, por favor consulte la sección “Uso de medidas no GAAP” anterior.
**Las ganancias por acción no GAAP se calculan utilizando acciones diluidas, mientras que la pérdida neta por acción no-GAAP se calcula utilizando acciones básicas, ya que todos los demás instrumentos son anti-dilutivos.
***La diferencia entre el promedio ponderado del número de acciones ordinarias utilizadas para calcular las ganancias por acción diluida GAAP y no GAAP durante los tres y nueve meses que finalizaron el 30 de septiembre de 2024 se debe a la exclusión del posible canje en acciones de los Bonos Convertibles 2027 en el cálculo GAAP, ya que su inclusión habría sido anti-dilutiva durante esos períodos.




Fuente: Sarepta Therapeutics, Inc.
_________________________________
1 Iff J, et al. Delayed Pulmonary Progression in Golodirsen-Treated Patients With Duchenne Muscular Dystrophy vs Mutation-Matched External Controls. Presentado en la MDA, 2024
2 Kuntz N, et al. Pulmonary Function in Advanced-Stage Patients With Duchenne Muscular Dystrophy Treated With Casimersen. Presentado en la WMS, 2025.
3 Iff J, , et al. Survival among patients receiving eteplirsen for up to 8 years for the treatment of Duchenne muscular dystrophy and contextualization with natural history controls. Muscle & Nerve. 2024; 70(1): 60-70. doi:10.1002/mus.28075.
4 Datos en el texto.
5 Mathews K, et al. Comparative Analysis of Loss of Ambulation in Eteplirsen-Treated Patients With DMD in the EVOLVE Study and Propensity Score–Weighted External Controls. Presentado en la MDA, 2025.
6 Muntoni F, et al. Comparing Ambulatory Outcomes of Golodirsen-Treated Patients vs Mutation-Matched External Controls. Presentado en la CNS, 2025.
7 Iff J, et al. Association Between Exon-Skipping Therapy With Eteplirsen and Cardiac Outcomes in Duchenne Muscular Dystrophy. Presentado en la MDA 2025.
8 Iff J, et al. Journal Comp Eff Res. 2023 Sep;12(9):e230086. doi: 10.57264/cer-2023-0086.
Contactos
Contacto con inversores
Ian Estepan, 617-274-4052, iestepan@sarepta.com
Ryan Wong, 627-800-4112, rwong@sarepta.com
Contactos para la prensa
Tracy Sorrentino, 617-301-8566, tsorrentino@sarepta.com
Kara Hoeger, 617-710-3898, khoeger@sarepta.com
Esta publicación está escrita en español para promover el acceso de la comunidad a información y actualizaciones. La información que se comparte tiene como objetivo el interés y la concientización general y, por lo tanto, no debe ser considerada ni interpretada como consejo médico o legal. Se trata de información publicada abiertamente. La Fundación Akari no promociona ninguna empresa, producto o tratamiento específico. Le recomendamos que consulte la fuente original y tome decisiones informadas.
Para leer la nota original de divulgación en inglés consulte:
Si desea saber más información acerca de este artículo de divulgación, contáctese directamente con la fuente original de esta nota.