Síguenos en Nuestras Redes Sociales
Por Annie Tang y Toshifumi Yokota*
Departamento de Genética Médica, Universidad de Alberta, 116 St. y 85 Ave., Edmonton, AB T6G 2R3, Canadá
*Autor a quien debe enviarse la correspondencia.
2025, 16(7), 777; https://doi.org/10.3390/genes16070777
Envío recibido: 26 de mayo de 2025 / Revisado: 25 de junio de 2025 / Aceptado: 26 de junio de 2025 / Publicado: 30 de junio de 2025
(Este artículo pertenece a la Sección Genómica Humana y Enfermedades Genéticas Human Genomics and Genetic Diseases)
Visitas al artículo: 544
Resumen
La distrofia muscular de Duchenne (DMD) es un desorden severo de desgaste muscular heredado que se asocia con morbilidad grave y mortalidad a nivel mundial. Las opciones de tratamiento actuales han mejorado la calidad de vida de los pacientes, pero estos tratamientos son sólo paliativos. Existe una necesidad de más opciones de tratamiento para la DMD. Las terapias de oligonucleótido antisentido (ASO, por sus iniciales en inglés) han emergido como una opción de tratamiento personalizada prometedora para grupos de pacientes que tienen mutaciones específicas. Un subgrupo de estas terapias puede omitir exones (que alteran el marco de lectura) en el gen DMD y puede restaurar parcialmente la producción de distrofina en personas con DMD. Una terapia de omisión de exón novedosa que está actualmente siendo investigada es brogidirsen, un exón 44 que apunta a la terapia ASO, utilizando un novedoso enfoque de doble objetivo. Este artículo proporcionará una revisión de la historia de brogidirsen y sus desarrollos en ensayos clínicos actuales. Resumirá cómo esta terapia en investigación se compara con otras terapias de ASO en etapa de ensayo preclínica y clínica, las cuales también apuntan al exón 44. También se discutirán los avances actuales y retos que la terapia basada en ARN enfrenta. En general, brogidirsen es una potencial incorporación prometedora a las opciones de tratamiento para la DMD existentes, con los resultados del ensayo clínico mostrando niveles de expresión por encima de la cantidad máxima de expresión de distrofina alcanzada por las terapias de omisión de exón para la DMD aprobadas por la Administración de Alimentos y Medicamentos de EE. UU. (FDA, por sus siglas en inglés) y la Agencia Europea de Medicamentos(EMA, por sus siglas en inglés). Se necesitará mayor investigación para determinar su eficacia general y habilidad para superar las limitaciones conocidas que otras terapias de ASO existentes enfrentan.
Palabras clave: distrofia muscular de duchenne; omisión de exón; exón 44; oligonucleótido antisentido; brogidirsen; ensayo clínico; distrofina; NS-089/NCNP-02; modulación de empalme; terapias antisentido
- Introducción a la Distrofia Muscular de Duchenne y Tratamientos
1.1 Patología de la Distrofia Muscular de Duchenne (DMD)
La DMD es un desorden neuromuscular progresivo relacionado con el cromosoma X que afecta a 19.8 de cada 100,000 hombres nacidos vivos a nivel mundial [1, 2]. Es causada por mutaciones en el gen DMD, lo que conduce a la falta de distrofina funcional, una proteína citoesquelética importante [3]. A diferencia de las mutaciones en el gen DMD que resultan en la distrofia muscular de Becker (DMB) más leve, la DMD es causada por la pérdida de función del gen DMD que alteran el marco de lectura de la traducción [4]. Esto resulta en la pérdida de distrofina funcional, lo cual hace que el tejido muscular funcional sea gradualmente reemplazado con tejido fibroadiposo [5]. La proteína distrofina en forma de varilla juega un papel esencial en el mantenimiento de la membrana de las células musculares al enlazar el citoesqueleto de actina subsarcolémico a la parte interior del sarcolema [3]. Una falta de distrofina interrumpe la señalización subcelular de miofibrillas, lo cual resulta en el incremento de estrés y daño en la membrana muscular, conduciendo en última instancia a la degeneración progresiva del esqueleto y del músculo cardiaco [6]. Esto resulta en la pérdida de ambulación y muerte eventual por complicaciones respiratorias o cardiacas entre los treinta y cuarenta años, incluso con los tratamientos de prolongación de vida actuales [7, 8]. Los tratamientos de esteroides estándar para la DMD apuntan a retrasar la progresión de la enfermedad, pero hay una necesidad no satisfecha de más tratamientos efectivos [9].
Las terapias paliativas actuales han extendido la esperanza de vida de algunos pacientes con DMD de haber tenido una historia natural sin tratar de entre 20 años hasta los 30-40 años con tratamiento [10]. Las cirugías de la columna vertebral y las tecnologías de soporte para ventilación nocturna han mejorado significativamente las tasas de sobrevivencia [11]. Aunque son efectivos en retardar la progresión de la enfermedad, los tratamientos con corticosteroides estándar tienen también efectos secundarios severos, como osteoporosis, cataratas, aumento de peso, y cambios en el comportamiento [11, 12]. A pesar de que estos efectos secundarios son monitoreados e incluídos en los cuidados a tener en cuenta, se requieren mayores avances en el área. Algunas estrategias actuales apuntan a restaurar los niveles de distrofina a través de la entrega de genes, los sistemas de entrega de CRISPR-Cas9, o de omisión de exón. Sólo se necesita 20% de la expresión de distrofina regular para reducir los síntomas más severos de la DMD y producir un curso más leve de la enfermedad [13]. Aún así, este nivel de restauración de distrofina en pacientes no se ha logrado con ninguna de las estrategias de tratamiento antes mencionadas, indicando la necesidad de esfuerzos continuos para mejorar las terapias para la DMD.
Los síntomas de la DMD son frecuentemente observados alrededor de los 3 a 4 años de edad, a través de manifestaciones críticas como la maniobra de Gower, marcha de pato, caídas frecuentes, torpeza, y marcha de puntillas [14]. Sin embargo, muchos casos pueden diagnosticarse antes a través del reconocimiento, examinación e investigación de cualquier infante masculino que no camina de manera independiente a la edad de entre 18 a 24 meses. Un diagnóstico debe ser confirmado tan pronto como sea posible para permitir asesoramiento genético para los padres y familia extendida. Un diagnóstico normalmente se confirma alrededor de los 5 años [15]. Las pruebas de diagnóstico incluyen electrocardiogramas, pruebas de creatina quinasa, pruebas genéticas, y biopsias del músculo [16]. Los niveles elevados de creatina quinasa normalmente se evalúan tempranamente, y las pruebas genéticas se usan para confirmar el diagnóstico al identificar la ubicación, tipo y la numeración en la escala de patogenicidad de mutaciones dentro del gen DMD en el exón 79 [17]. Alrededor del 70% de pacientes con DMD tienen deleción (es) o duplicación (es) cubriendo uno o múltiples exones en el gen DMD, lo que puede ser rápidamente detectado usando pruebas genéticas [14]. Las biopsias de los músculos se usan luego para confirmar la ausencia de distrofina [14]. De manera interesante, las portadoras femeninas de DMD pueden también manifestar creatina quinasa elevada [18]. Esto usualmente representa una distribución desplazada de los niveles de creatina quinasa comparados con el rango normal, que puede ser marcadamente incrementado en algunos portadores manifestantes clínicamente sintomáticos. Esta enfermedad afecta la ambulación, causando dificultades al brincar, correr, subir escalones, y levantarse del piso [2, 14]. Los planes de cuidado y tratamiento cambian entre las etapas ambulatorias y no ambulatorias de la enfermedad, pero el manejo de la rehabilitación y el uso de glucocorticoides siguen siendo elementos esenciales a lo largo de toda la vida [14].
La severidad de la DMD empeora con la edad, incluso con estrategias farmacológicas, quirúrgicas y holísticas actuales [19]. La escoliosis, cardiopatía, y problemas respiratorios usualmente empiezan a desarrollarse en los años adolescentes [19]. La aparición de escoliosis y la pérdida de ambulación normalmente ocurre alrededor de los 10-14 años [20, 21]. Algunos pacientes con DMD también experimentan deterioro cognitivo comórbido o condiciones del comportamiento, incluyendo síndrome del espectro autista, síndromes obsesivos-compulsivos, ansiedad, discapacidad intelectual, o síndrome de déficit de atención e hiperactividad [22]. Alrededor de la mitad de los pacientes con DMD tienen una discapacidad de aprendizaje [23]. Esta enfermedad ha sido asociada con interrupciones del sueño, problemas gastrointestinales, dolor crónico, rigidez, problemas al masticar, obesidad, enfermedad cardíaca, y problemas de salud mental [24]. La carga epidemiológica y la frecuencia de la DMD no se relacionan a ningún aspecto demográfico, con poblaciones diferentes generalmente teniendo niveles similares de riesgo. Sin embargo, las diferencias de edad en el diagnóstico y planes de tratamiento varían globalmente y entre los distintos aspectos demográficos [25]. Los costos económicos, sociales, y uso de la atención sanitaria asociados con la DMD se elevan conforme la enfermedad progresa, haciendo que la morbilidad y mortalidad de la condición establezcan una carga significativa en los sistemas de salud a nivel mundial [26].
1.2 Tratamientos antisentido para la DMD
Los oligonucleótidos antisentido (ASOs) son medicamentos análogos basados en ácidos nucleicos sintéticos que han mostrado potencial terapéutico. Los ASOs actuales aprobados por la FDA usados para tratar la DMD y otras enfermedades incluyen viltolarsen, casimersen, golodirsen, eteplirsen, nusinersen, tofersen, y muchos otros. Cuando se usan para tratar la DMD, las terapias de ASO apuntan a restaurar la producción de distrofina funcional parcialmente para reducir la degeneración muscular y retrasar la progresión de la enfermedad [27]. Sarepta se ha orientado a diferentes exones del gen DMD al desarrollar eteplirsen (exón 51), golodirsen (exón 53), y casimersen (exón 45) [28, 29, 30]. Nippon-Shinyaku y NS Pharma han avanzado con vitolarsen (exón 53) al hacerlo tratamiento aprobado por la FDA y actualmente están llevando a cabo ensayos clínicos con brogidirsen (exón 44) [31, 32].
Un subgrupo de terapias de ASO apuntan a usar la omisión de exón para retrasar la progresión de la DMD. Al haber mutaciones patógenas de ADN dentro y fuera del marco, la omisión de exón podría requerir que más de un exón sea omitido para regresar a la lectura en el marco del ARN transcrito [33]. Las terapias de ASO de omisión de exón pretenden omitir segmentos específicos, conocidos como exones, del gen DMD que son mutados. Esto restaura el marco de lectura abierto del gen DMD, lo cual puede conducir a la producción de una versión truncada de distrofina [34]. Estas proteínas distrofinas acortadas son parcialmente funcionales y se piensa que pueden contribuir al complejo de distrofina-glicoproteína para proporcionar soporte estructural y estabilidad al sarcolema. Estas terapias de omisión de exón se orientan al pre ARNm, una etapa donde el ARNm aún no está listo para la traducción en proteína distrofina. Estos no necesariamente apuntan a la región mutada del gen DMD, pues en lugar de eso pueden orientarse a las partes de flanqueo que inducen la omisión de aquellas partes mutadas. [35]. El objetivo general es convertir el fenotipo DMD en un fenotipo DMB más leve al usar ASOs para alterar el empalme del pre ARNm de modo que resulte en la producción de una proteína distrofina parcialmente funcional [34].
Los exones del gen DMD pueden ser uno de los nucleótidos 3n, 3n+1, ó 3n-1; es por esto que las deleciones de exón, el tipo más común de mutación de distrofina [33], pueden tener tres efectos diferentes en el marco de lectura. Añadir una ‘deleción’ usando la omisión de exón puede sólo corregir el marco de lectura si la ‘eliminación’ complementa una deleción fuera del marco existente. Por ejemplo, un tipo 3n+1 de deleción sería capaz de restaurar el marco de lectura sólo para ciertas mutaciones. Inducir la deleción del exón 44, en este caso, puede entonces sólo compensar las deleciones del tipo 3n-1. Esto se representa con diferentes formas de exón en diagramas, con algunas deleciones de omisión de exón potenciales resultando en dos exones que no encajan juntos en un marco de lectura funcional (Figura 1). Estos son factores a considerar al diseñar terapias de omisión de exón de ASO, junto con la eficiencia de la omisión del exón, la funcionalidad de la proteína, y la aplicabilidad potencial para otras mutaciones [33].
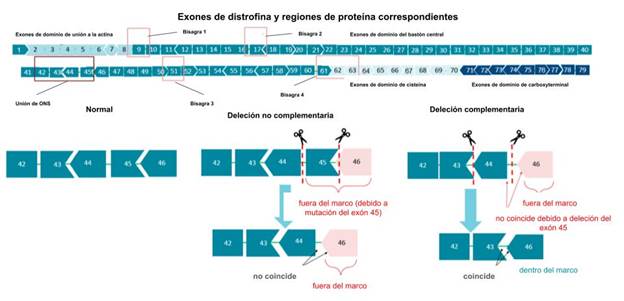
Figura 1. Los exones distrofina y el efecto de diferentes deleciones de exón en el marco de lectura del ARN. Los exones de diferentes formas representan cuáles exones “encajan” juntos para restaurar un marco de lectura funcional. El gen DMD tiene muchos exones que pueden ser objeto de los ASOs. En pacientes con DMD, las deleciones frecuentemente se encuentran en un punto mutacional 3’ entre los exones 43 y 55, con algunos estudios limitando la región a los exones 45 a 55 [36, 37]. Las deleciones de los exones 43, 45, y 52 son comunes [38]. Se ha identificado otro punto mutacional dentro de los exones 3 al 9 y se conoce como el punto mutacional 5’ [39]. Los ASOs de omisión de exón están diseñados para restaurar el marco de lectura del ARNm al omitir las regiones mutadas. Estos ASOs pueden usarse para tratar pacientes con deleciones, duplicaciones, o pequeñas mutaciones en el gen DMD [40]. La omisión de un solo exón es más aplicable a pacientes con deleciones en el dominio de bastón de distrofina [40]. Esta región de dominio de proteína es más tolerante a las deleciones internas porque contiene varios dominios repetidos similares a la espectrina y regiones bisagra [2]. Al orientarse a mutaciones específicas dentro de la DMD, las terapias de ASO ofrecen una opción de tratamiento personalizada para pacientes con mutaciones elegibles y susceptibles.
Los ASOs pueden ser clasificados estructuralmente en Ácido Nucleico Bloqueado (LNA, por sus iniciales en inglés), estructura de ARN con modificación 2’-O-metil (MOE, por sus iniciales en inglés), y agrupaciones de subclase de oligómero morfolino fosforodiamidato (PMO, por sus iniciales en inglés) [41]. Las moléculas de LNA se bloquean en una sola conformación porque contienen un puente entre los fragmentos moleculares de azúcar y la columna vertebral de fosfato o el anillo de furanosa [42]. Las moléculas modificadas de MOE son más resistentes a nucleasas que el ARN no modificado, principalmente debido a que sustituyen un 2’-etileno glicol por la ribosa 2’OH que normalmente se encuentra en el ARN [43]. Actualmente, las terapias de PMO han dominado el panorama para la DMD aprobado por la FDA y han sido más desarrolladas que las terapias para la DMD basadas en LNA o MOE. Los cuatro ASOs tratantes aprobados condicionalmente para la DMD (eteplirse, vitolarsen, casimersen, y golodirsen) son PMO. [44]. Estas terapias de PMO difieren del ARN/ADN regular a través de modificaciones de azúcar y fosfato. Tienen una columna vertebral hecha de estructuras en forma de anillos de morfolino en lugar de la columna vertebral de ribosa regular. Estos anillos de morfolino están entonces conectados a través de enlaces de fosforodiamidato en lugar de las enlaces regulares cargados con inter-nucleósido de fosfodiéster [45]. La síntesis de PMOs empieza con la apertura de los anillos de ribosa de cinco miembros dentro de ribósidos usando la oxidación [46]. Estos anillos se cierran después usando amoniaco para generar un anillo sustituto de morfolino de seis miembros [46]. Los enlaces de fosforodiamidato se añaden después para reemplazar los enlaces de fosfodiéster [46]. Estos enlaces de fosforodiamidato resultan en alta estabilidad metabólica, resistencia enzimática, y solubilidad acuosa media [47]. Los PMOs también carecen de un grupo carbonilo usualmente presente en los ácidos nucleicos, lo cual confiere resistencia contra ataques por proteasas, nucleasas, y esterasas en sueros biológicos [48]. Estas moléculas son también altamente resistentes a hidrolasas [48]. Estas propiedades resultan en algunas interacciones con moléculas biológicas en general, pero también en una habilidad para unir fuertemente y poder orientarse a las secuencias [49].
Los PMOs se consideran tratamientos prometedores para varias enfermedades por su habilidad para incrementar o disminuir la expresión génica a través de un impedimento estérico o mecanismo de modulación de empalme [49, 50]. Estos tienen cargas neutrales y se usan de manera extensiva para aplicaciones génicas antisentido en cáncer y biología evolutiva [45]. Los tratamientos con ASO para la DMD pueden provocar un mecanismo de modulación de empalme para rescatar la expresión génica [51]. Los PMOs pueden unirse a secuencias complementarias a través de emparejamiento de base Watson-Crick orientándose a las secuencias de pre-ARNm ya sea cerca o en uniones exón-intrón para alterar el empalme del ARNm [52]. Esto puede excluir exones con mutaciones que interrumpen el marco de lectura y resultan en la producción de una versión interna de distrofina truncada de los genes propios del paciente. Otros mecanismos, como la terapia génica y mecanismos de entrega CRISPR-Cas9, existen para restaurar la función también pero, ya sea que no hayan progresado pasando la etapa pre-clínica, o que estén actualmente atravesando evaluaciones para su eficacia y efectos inmunogénicos [53]. Las reacciones inmunogénicas para las terapias de ASO eran una preocupación mayor para la primera generación de ASOs, pero las modificaciones estructurales han disminuido los riesgos de inmunogenicidad para los tratamientos actuales con ASO [54]. Estas mejoras han acentuado el potencial terapéutico y perfil de seguridad de las terapias de ASO, apoyando su desarrollo continuo como un enfoque viable para tratar desórdenes genéticos como la DMD.
Este texto resumirá información sobre ello y presentará una comparación entre brogidirsen (NS-089/NCNP-02) y otras terapias de omisión del exón 44 en desarrollo. Se centrará en el potencial terapéutico de la omisión del exón 44 y se remontará a desarrollos recientes para candidatos a omisión de exón en el área terapéutica de oligonucleótidos para la DMD. A pesar del estrecho margen de este texto, la comparación que se presenta es útil debido a que ilustra cómo ciertos aspectos de diseño pueden influenciar los atributos clínicos de la medicina oligonucleótida. El diseño de una sola secuencia de PMO lineal que se orienta a dos reguladores discretos internos de empalme con el exón 44 parece añadir suficiente potencia al inicio de cualquier posible necesidad para la conjugación de un anticuerpo objetivo o péptido penetrador de célula, en el sentido de que es realizado con la otra omisión del exón 44 en desarrollo clínico.
- Terapias de omisión del exón 44
2.1 Mecanismos y objetivos
Actualmente, no hay terapias de omisión del exón 44 aprobadas federalmente. Las terapias de omisión mediadas por oligonucleótido que apuntan al exón 44 tienen el potencial de tratar hasta el ~7% de pacientes con DMD [55]. Esto es debido a su habilidad para abordar mutaciones que frecuentemente se concentran en regiones alrededor del exón 44 del gen DMD. Extenderse del exón 43 al 55 es un punto mutacional dentro del gen DMD, con la eliminación más común en el gen DMD siendo el exón 45 [34, 37]. Dado el número relativamente reducido de pacientes con DMD con deleciones del exón 44-45 y una relación entre esta deleción y un fenotipo más leve, se cree que la omisión del exón 44 en niños con DMD que ya tienen una deleción del exón 45 u otra deleción susceptible a omisión del exón 44 resulta en la restauración del marco de lectura y brinda una proteína distrofina acortada pero parcialmente funcional [34]. El personal clínico identifica pacientes candidatos que son susceptibles a la omisión del exón 44 a través de genotipificación. La omisión del exón 44 puede ser aplicable terapéuticamente a pacientes con deleciones comprendiendo los exones 45-54, sólo el exón 45, un subgrupo de deleciones finalizando en el exón 43, y muchas otras formas de deleciones [34]. Aunque no todas las deleciones finalizando en el exón 43 son susceptibles, las que son aplicables incluyen algunas deleciones de los exones DMD 14-43, 19-43, 30-43, 35-43, 36-43, 40-43, y 42-43 [34, 40]. La meta de estas terapias es facilitar la producción de una proteína distrofina truncada que retenga la función parcial, como la que se encuentra presente en algunos pacientes con DMB.
Varios ASOs que apuntan al exón 44 DMD están actualmente en la etapa de ensayo clínico. Avidity Biosciences está desarrollando AOC1044, Entrada Therapeutics ha avanzado con ENTR-601-44, y NS Pharma está llevando a cabo ensayos clínicos para brogidirsen (NS-089/NCNP-02). Actualmente, el candidato de NS Pharma es el que más ha avanzado en la línea, habiendo sido sometido a ensayos de Fase I/II o II en pacientes con DMD. Sarepta Therapeutics, Wave Life Sciences, PepGen, y Dyne Therapeutics también están desarrollando terapias de omisión de exón que actualmente se encuentran en etapa pre-clínica. Estos esfuerzos se detallarán a continuación y reflejan una línea en crecimiento de la próxima generación en terapias de omisión de exón que integran diseños novedosos de oligonucleótido y plataformas de entrega que pretenden mejorar la absorción del tejido y eficiencia de la omisión de exón.
2.2 Resultados preclínicos y clínicos actuales de AOC1044 y ENTR-601-44
AOC1044, también conocido como delpacibart zotadirsen, por Avidity Biosciences, es una terapia de omisión del exón 44 que consiste en cuatro copias PMO adjuntas a un anticuerpo monoclonal humanizado receptor 1 de anti-transferrina (TfR1) a través de enlazadores [56, 57]. La conjugación anticuerpo-PMO es para dirigir mejor los PMOs al tejido muscular, en el sentido de que el objetivo TfR1 es expresado en todas partes en las membranas de las células esqueléticas, cardíacas, y las más suaves de las musculares. [58]. En modelos preclínicos humanizados de ratón mdx de DMD con una deleción del exón 45 se ha observado que esta terapia restaura alrededor del 20% de la distrofina en el músculo esquelético y 6% en el músculo cardiaco [59]. Los niveles de restauración de distrofina en el músculo cardíaco son particularmente interesantes. Esto es porque las terapias existentes son ineficientes al restaurar distrofina en el tejido cardíaco, un área que necesita ser enfocada para reducir los efectos de la miocardiopatía [59, 60]. La seguridad y tolerabilidad de esta terapia fueron probadas en ensayo clínico de Fase I/II aleatorizado, doble ciego, controlado con placebo EXPLORE44 en 40 humanos sanos voluntarios (NCT05670730). Los resultados mostraron que el grupo tratado con AOC1044 sólo presentó eventos adversos de leves a moderados, indicando que la terapia fue bien tolerada [56]. También se observaron incrementos dependientes de la dosis en eficiencia de omisión del exón después de administrar una dosis de 5 mg/kg ó 10 mg/kg [56]. Actualmente, esta terapia está siendo probada en pacientes con DMD en la segunda parte del ensayo EXPLORE44.
ENTR-601-44 consiste en un PMO de omisión del exón 44 conjugado con un péptido patentado que sirve de vehículo de escape endosomal (EVV, por sus iniciales en inglés). Entrada Therapeutics tiene múltiples terapias conjugadas en desarrollo, y ha optimizado la tecnología EEV para mejorar la eficiencia de entrega intracelular en tejidos y órganos específicos [44, 61]. El EEV contiene péptidos que penetran a la célula que inducen la gemación y colapso de endosomas [56]. Esto ayuda a ENTR-601-44 a escapar del atrapamiento endosomal e incrementa su efectividad [63]. Los estudios preclínicos en ratones D2-mdx mostraron omisión de exón en tejido esquelético y cardiaco [64]. En el Reino Unido, se examinaron la seguridad y tolerabilidad de ENTRE-601-44 en voluntarios masculinos sanos de edades 18 a 55 de agosto de 2023 a octubre de 2024 en un estudio controlado con placebo (ISRCTN36174912). Cuatro niveles de dosis fueron puestos a prueba, con exámenes regulares de sangre y orina para monitorear la función renal y los niveles de plaquetas [65]. Esta terapia tuvo una suspensión clínica por la FDA de dos años que se quitó en febrero de 2025 y comenzará a evaluarse en Estados Unidos en el ensayo ELEVATE-44-102, un estudio clínico de múltiples dosis ascendientes de Fase 1b, aleatorizado, doble ciego, controlado con placebo [61]. Está acordado empezar este ensayo en 2026 y administrará ENTR-601-44 en dosis que van de 0.16 mg/kg a 1.28 mg/kg, en alrededor de 32 pacientes con DMD ambulatorios y no ambulatorios [61].
- Estudios y ensayos de Briogidirsen
3.1 Resultados de estudios preclínicos
Briogidirsen (NS-089/NCNP-02) es un PMO sin modificar con un enfoque novedoso de doble objetivo para el exón 44 de DMD [32, 66]. Esta terapia obtuvo la designación de medicamento huérfano y el estatus de terapia innovadora en Estados Unidos y Europa durante 2023 [67]. La secuencia de 24 ácidos nucleicos de NS-089/NCNP-02 es ‘TTGTGTCTTTCTTCTGTTAGCCAC’ (Código FDA UNII: 4KM8P975UM). Brogidirsen contiene dos secuencias de 12 bases conectadas directamente que están diseñadas para omitir el exón 44 al dirigirse a las posiciones 64-75 y 92-103 dentro del exón (Figura 2) [68]. Se ha mostrado potencial terapéutico en un estudio preclínico previo, donde un tratamiento de incubación de 1 hora con fibroblastos derivados de pacientes y convertidos con MyoD, presentando una deleción del exón 45 del gen DMD, resultó en una omisión de exón 44 con una EC de 0.63 μmol/L 7 días después de la incubación con el tratamiento de NS-089/NCNP-02. En un ensayo clínico posterior por Nippon-Shinyaku y NS Pharma (NCT04129294), se observó que el tratamiento restauró la expresión de distrofina a aproximadamente el 25% de los niveles observados en controles saludables [66, 68]. Una vez que NS-089/NCNP-02 se une a los lugares dirigidos en el ARN, los exones que cambian de marco pueden ser empalmados y omitidos (Figura 3). Debido a que las PMOs carecen de carga, se percibe que tienen un mejor perfil de seguridad y estabilidad mejorada en comparación con los ASOs cargados [44, 69].
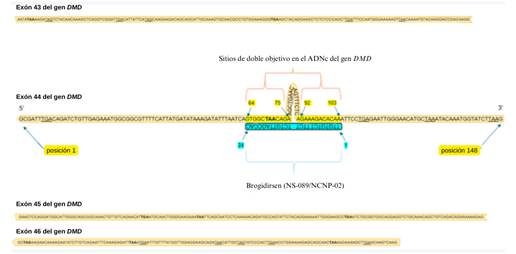
Figura 2. Los sitios orientados de brogidirsen (NS-089/NCNP-02) dentro de la secuencia del exón 44 del gen DMD. Las letras negritas y lo resaltado indican codones de parada.
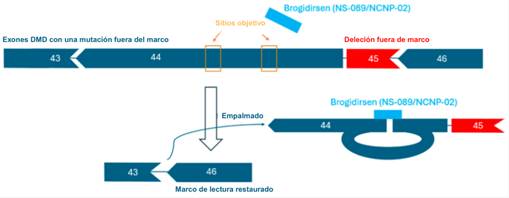
Figura 3. Mecanismo de restauración del marco de lectura de brogidirsen. Los dos sitios dentro de brogidirsen apuntan a secuencias dentro del exón 44 del gen DMD.
De manera preclínica, NS-089/NCNP-02 se identificó de entre varios ASOs individuales que contenían dos secuencias apuntando al exón [70]. De los 26 PMOs de 22 bases, se observaron niveles elevados de omisión del exón 44 para las moléculas apuntando a las posiciones 11-52 y 61-122 [70]. A través de una detección más detenida, los oligómeros apuntando a las posiciones 21-33, 61-73, y 91-103 mostraron tener actividad elevada de omisión [70]. Se identificó que los candidatos de 22 bases y 26 bases tienen los niveles más altos de actividad, y la molécula de 24 bases se seleccionó para mayor estudio [70]. Esta molécula de 24 bases fue NS-089/NCNP-02, y la prueba de NS-089/NCNP-02 en células humanas de rabdomiosarcoma mostró un incremento dependiente de la dosis en la eficiencia de omisión de exón cuando se midió a través de RT-PCR [70].
Este tratamiento se puso a prueba después en miotubos DMD derivados de pacientes con una deleción en el exón 45 que era susceptible a la omisión del exón 44 [70]. Se observaron los niveles aumentados de la expresión de proteína distrofina, pues se vieron niveles de expresión de distrofina sin DMD de 4.7 %, 5.1%, 14.0%, 13.5%, 21.8%, 19.3% y 38.0% después del tratamiento con concentraciones de μmol/L de 0.01, 0.03, 1, 3, y 10 de NS-089/NCNP-02, respectivamente [70]. Un estudio de incubación de 7 días también se llevó a cabo en miotubos DMD derivados de pacientes. Los resultados mostraron que la concentración efectiva semi-máxima, o EC50, de NS-089/NCNP-02 fue de 0.63 μmol/L [70]. Como los PMOs tienen relativamente una vida corta de 2 horas en el torrente sanguíneo, el propósito de este estudio fue determinar si los PMOs tomados por células en ese corto periodo pudieran ser efectivas y exhibir actividad [71]. Se encontró que la actividad de la omisión del exón 44 persistió por al menos una semana tras el tratamiento con NS-089/NCNP-02 [70]. Los resultados después de 4 semanas de administraciones intravenosas a monos cinomolgos también mostraron potencial con omisión del exón observada en dosis de 66, 220, y 660 mg/kg por semana [66, 70]. En cambio, cuando se trató por 13 semanas, la omisión del exón se observó en el músculo cardiaco en dosis de 600 mg/kg o mayores [70]. Las eficiencias de la omisión del exón de hasta 16.2 % fueron observadas en el músculo esquelético en la dosis más alta de 2000 mg/kg [70]. Aunque la omisión del exón se observó en el miocardio, estas diferencias en el músculo del miocardio de los monos no fueron significativas, con la dosis más alta resultando en una eficiencia de omisión de 0.8%, Estos resultados in vitro y en vivo llevaron a NS Pharma a iniciar un estudio de fase I/II para NS-089/NCNP-02.
3.2 Hallazgos Clínicos y Farmacología
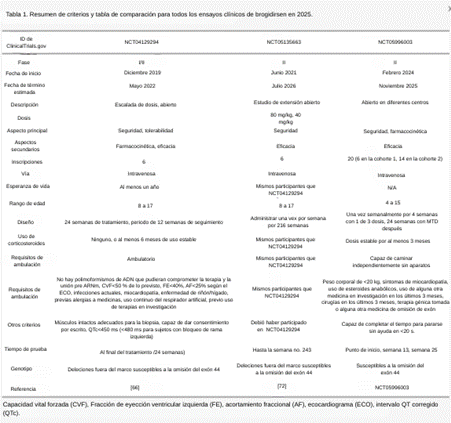
Se han registrado tres ensayos clínicos para NS-089/NCNP-02 (Tabla 1). Los resultados del primer ensayo clínico se publicaron en 2025 [66]. Con duración de 2019 a 2022, a seis pacientes con DMD ambulatorios, de edades entre 8 y 17 años con deleción del exón 44 susceptible a omisión de exón se les administró NS-089/NCNP-02 semanalmente por dos semanas en una de las cuatro dosis diferentes, en rangos de 1.62 a 80 mg/kg por semana (NCT04129294). Este estudio se orientó a evaluar la seguridad, farmacocinética, y eficacia del tratamiento de omisión de exón. Esta terapia fue tolerada de manera general a lo largo de todos los niveles de dosis (1.62, 10, 40, y 80 mg/kg por semana) en escalada de dosis la mitad del estudio. Las biopsias de los músculos bíceps se realizaron previas al tratamiento y después de un periodo de 24 semanas de tratamiento para medir los niveles de distrofina usando electrotransferencia e inmunohistoquímica [66]. Se usó espectrometría de masas y cromatografía líquida para evaluar la farmacocinética en las muestras de sangre obtenidas en diferentes momentos previos al tratamiento y posteriores al tratamiento en el día 1, así como en el día 2, día 8, día 9, día 15, y día 22, durante la primera parte del estudio. Durante la segunda parte del estudio, las muestras de sangre se tomaron en diferentes momentos del día 1 antes y después de la administración, junto con recolecciones en el día 2, día 8, día 22, día 50, día 78, día 106, día 134, día 162, día 163, y ya fuera en el día 169 o día 176 [72]. El análisis de orina utilizó orina acumulada en los días 2, 4, y 11 para la primera parte del estudio, y en los días 2 y 163 para la segunda parte del estudio [72]. Esta terapia fue bien tolerada de manera general a lo largo de los niveles de dosis (1.62, 10, 40, y 80 mg/kg por semana) en la primera escalada de dosis durante la mitad del estudio. No se observaron toxicidades que limitaran la dosis o anticuerpos anti distrofina [66]. Los eventos adversos más frecuentes fueron niveles de microgobulina β2, proporciones de albúmina/creatinina, y niveles de cistatina C en la orina incrementados [66]. Hubo una ausencia de toxicidad hepática notable en las dosis evaluadas, indicando un perfil de seguridad similar a otros PMOs [66]. De manera general, la ausencia de eventos adversos serios o discontinuaciones relacionadas con eventos adversos respalda un perfil de seguridad tolerable en el rango de dosis evaluada [66].
Tabla 1. Resumen de criterios y tabla de comparación para todos los ensayos clínicos de brogidirsen en 2025.
El segundo parámetro de este estudio fue la eficacia, la cual se midió a través de evaluaciones funcionales y examinando la biopsia del músculo para los niveles de expresión de proteína distrofina. Los niveles de creatina quinasa en el suero disminuyeron a lo largo del periodo de tratamiento de 24 semanas en los participantes que recibieron dosis de 40 y 80 mg/kg por semana. El desempeño funcional motor se midió usando la Escala Ambulatoria North Star, la prueba para caminar 10 metros, el tiempo para pararse, las pruebas para caminar 6 minutos y 2 minutos, la prueba de tiempo para subir 4 escalones, la evaluación funcional de extremidades superiores, prueba de fuerza de agarre en la mano, y evaluaciones de fuerza muscular (abducción del hombro, flexión/extensión de la rodilla, y flexión/extensión del codo) [66]. El desempeño funcional disminuyó en las dos cohortes de dosis baja (1.62 y 10 mg/kg por semana) y se estabilizaron con una tendencia hacia la mejora en las cohortes de dosis alta (40 a 80 mg/kg por semana) [66]. Debido a que la DMD es una condición progresiva, los pacientes entre las edades de 8 y 17, normalmente experimentan un declive en la función física según estudios de historia natural [73]. De aquí que el desempeño estabilizado en las pruebas de función motora mencionadas arriba indique que hay una posibilidad para este tratamiento de estabilizar la función muscular a largo plazo, indicando una necesidad de estudios clínicos a largos plazos para esta terapia en investigación.
Según los análisis no clínicos e in silico, hay potencial para NS-089/NCNP-02 de unirse a productos de ARN de genes aparte del gen DMD, lo cual resulta en lo que conocemos como efectos fuera del objetivo o no deseados. Estos análisis encontraron que NS-089/NCNP-02 puede potencialmente unirse al exón 8 del gen de fosfodiesterasa 3B para producir una proteína en estado no nativo potencialmente inmunogénica que pudiera resultar en inflamación y fiebre [66]. NS-089/NCP-02 pudiera también unirse al ARN del canal de cationes de la subfamilia M del receptor potencial transitorio, miembro 3, lo cual pudiera afectar la presión sanguínea [66]. Aunque estos análisis sugieren interacciones fuera del objetivo o no deseadas, no se observaron eventos adversos correspondientes durante el ensayo. No se observaron casos de fiebre y, de manera general, los marcadores inflamatorios se mantuvieron similares a aquellos antes del tratamiento para la mayoría de los pacientes [66]. Sin embargo, los niveles de IL-6 sí incrementaron en la semana 12 y luego disminuyeron en la semana 24 para un participante, y un incremento y disminución similares en TNF se presentó en otro [66]. Los niveles de IFN-y siguieron el mismo patrón en un tercer participante [66]. Estos marcadores inflamatorios sugieren una inflamación leve, pero es importante notar que los pacientes con DMD frecuentemente están en un estado inflamado de manera crónica por la constante degeneración muscular [74]. La inflamación puede tanto estimular la regeneración, como empeorar el daño muscular [74]. Los cambios en la presión sanguínea no se reportaron clínicamente al momento en que se redactó, pero los estudios preclínicos con monos no observaron efectos en la presión sanguínea [66].
En el ensayo clínico inicial para NS-089/NCNP-02, la eficiencia de la omisión del exón 44 se midió antes y después del tratamiento con NS-89/NCNP-02. Como se mencionó previamente, el ensayo clínico evaluó ambos resultados clínicos y los niveles de proteína distrofina de la biopsia en un periodo de 24 semanas, e involucró a seis pacientes con DMD (8-17 años de edad), quienes recibieron la medicina a través de inyección intravenosa (Tabla 1). Se usaron la electrotransferencia e inmunohistoquímica para probar los niveles de distrofina de las biopsias del músculo bíceps braquial hechas previas al tratamiento y después de 24 semanas de tratamiento [66]. Los resultados mostraron incrementos de distrofina dependientes de la dosis de 16.63% y 24.47% después de 24 semanas de tratamiento con dosis de 40 mg/kg y 80 mg/kg de NS-089/NCNP-02 [66]. La dosis alta (80 mg/kg) fue la más efectiva al restaurar los niveles de distrofina después de 24 semanas. Los datos del suero proteómico mostraron incrementos en la distrofina a las 12 y 24 semanas. Las concentraciones de NS-089/NCNP-02 de plasma y urinario también se midieron para evaluar la eficacia y se usaron para calcular los parámetros farmacocinéticos, como el rango de excreción urinaria [66]. Un estudio de extensión se llevó a cabo luego del ensayo de Fase I/II inicial y se espera que se complete en 2026 (NCT05125663). Dado que se ha teorizado que el 20% de restauración de distrofina es suficiente para reducir los síntomas de DMD más severos, la restauración de alrededor del 25% de los niveles de control sanos de distrofina es prometedora [13]. Este nivel de expresión sugiere que hay potencial para mejoras funcionales a largo plazo, aunque se necesitan estudios adicionales a largo plazo para confirmar este hallazgo.
Actualmente, NS Pharma está reclutando para un estudio de Fase 2 abierto en varios centros de NS-089/NCNP-02 (NCT05996003). Este estudio principalmente tiene por objetivo la seguridad, tolerabilidad, y farmacocinética del tratamiento, teniendo como objetivos secundarios los relacionados con la eficacia del tratamiento (Tabla 1). Las medidas del objetivo principal serán el número de eventos adversos, los parámetros de farmacocinética en el plasma de orina y sangre, y los resultados de electrotransferencia para observar los cambios en los niveles de distrofina en el músculo esquelético entre el inicio y la semana 25 [32]. Las medidas del objetivo secundario usarán la espectrometría de masas, tinción por inmunofluorescencia, prueba de fuerza de agarre en la mano, la Escala Ambulatoria North Star, el tiempo para caminar 10 metros, el tiempo para pararse, la distancia total para caminar por 6 minutos, el tiempo para subir 4 escalones, y la evaluación funcional de extremidades superiores [32]. Se dividirá en dos partes con dos cohortes. Los participantes serán ambulatorios, de edades entre 4 y 15 años, deberán tener mutaciones confirmadas que sean susceptibles a la omisión del exón 44, y deberán tener dosis estables de glucocorticoides. Se les administrará por vía intravenosa una dosis de NS-089/NCNP-02 semanalmente. La primera cohorte pasará por un plan de tratamiento de 4 semanas en uno de los tres niveles de dosis diferentes; luego, todos los participantes cambiarán al nivel de una sola dosis por un periodo de 24 semanas. El nivel de una sola dosis será la dosis máxima tolerada (MTD, por sus iniciales en inglés) con efectos secundarios aceptables. La Cohorte 2 pasará por un periodo de tratamiento de 24 semanas con la MTD determinada en la primera parte del estudio. Los pacientes con síntomas de miocardiopatía, o aquellos que hayan recibido terapia génica u otro tratamiento con ASO, serán excluidos. Este estudio intervencional se espera concluir cerca de finales de 2025.
De manera general, los ensayos completados y los ensayos clínicos en curso para NS-089/NCNP-02 han indicado un perfil de seguridad tolerable y han proporcionado niveles de restauración de distrofina en dosis más altas. Aunque se observó estabilización funcional en el ensayo clínico completado, la eficacia a largo plazo y la farmacocinética se mantienen como un área para estudio futuro. Los riesgos fuera del objetivo o no deseados también garantizan el monitoreo continuo de la presión sanguínea y las medidas de suero. El ensayo de Fase I/II de NS-089/NCNP-02 contó también con una población pequeña bajo estudio, y los ensayos futuros deberían probar la terapia en una cohorte de pacientes con DMD más grande. Si el siguiente ensayo clínico muestra un perfil de seguridad favorable, eficacia continua, y estabilización funcional en los pacientes, NS-089/NCNP-02 podría convertirse en un candidato clave para la terapia de omisión del exón 44 en la DMD.
- Comparación de Briogidirsen con otras terapias que apuntan al exón 44
TantoAOC 1044 como ENTR-601-44 tienen conjugaciones que tienen por objetivo mejorar la eficacia de los PMOs adjuntos al optimizar la biodisponibilidad o captación celular. De manera similar a ENTR-601-44, PGN-EDO44 usa la plataforma de PepGen de péptidos penetrantes celulares para oligonucleótidos de suministro mejorado [75]. Este péptido debe incrementar la entrega y absorción celular por los tejidos, además de ser usado con varios ASOs terapéuticos bajo desarrollo por PepGen [76]. De manera pre-clínica, los niveles de omisión del exón 44 promedio del 93.4% fueron observados después del tratamiento con PGN-EDO44 con la dosis más alta en el tipo de mioblastos humanos más fuerte [75]. PepGen pretende continuar el desarrollo de PGN-EDO44, junto con sus otros candidatos de omisión de exón. Otro péptido penetrante celular prometedor y de conjugación de PMO es DG9-PMO, el cual ha inducido el 47% de expresión de distrofina en tejidos cardiacos [77]. Estas conjugaciones muestran una tendencia hacia la siguiente generación de terapias de omisión de exón que combinan la química de oligonucleótidos optimizada con sistemas de entrega o suministro avanzados, teniendo por objetivo mejorar los resultados en pacientes con DMD a través de perfiles farmacocinéticos mejorados con orientación y captación de tejido reforzadas.
Dentro de esta tendencia en contexto, es notable que NS-089 no contiene conjugaciones con ningún péptido y aún recibe un porcentaje relativamente alto de restauración de distrofina de alrededor de 25% en pacientes con DMD. Debido a que carece de conjugaciones de péptido, NS-089/NCNP-02 confía en la biodistribución natural y receptores para encontrar y entrar en las células del músculo esquelético y cardiaco. Aunque estos niveles de omisión de exón son prometedores, si este efecto se debe solamente al enfoque de doble objetivo u otros factores – como la duración relativamente corta del tratamiento de 24 semanas, los diferentes niveles de dosificación, y el modesto número de pacientes– se necesitará realizar mayor estudio. El enfoque de doble objetivo de NS-089/NCNP-02, usando dos secuencias enlazadas enfocando diferentes puntos dentro del mismo exón, difiere de las secuencias de una orientación dentro de las terapias con AOC1044 y ENTR-601-44. Este es un enfoque novedoso no sólo en el campo terapéutico para la DMD, sino también para las medicinas ASO en general. Un estudio previo evaluó la entrega de dos ARNmi con diferentes puntos objetivos simultáneamente para terapéuticos de cáncer en investigación, pero ningún estudio, al momento en que se redacta, ha conjugado dos diferentes secuencias orientadas de ASO juntas, incluso en contextos de otras enfermedades. [66,78].
Actualmente, NS-089/NCNP-02 ha avanzado más clínicamente, que AOC1044 y ENTR-601-44 en las pruebas de terapias en pacientes con DMD. NS-089/NCNP-02 está listo para empezar un ensayo de Fase II más extenso luego de un ensayo de Fase I/II en seis pacientes con DMD, mientras que tanto AOC1044 y ENTR-601-44 actualmente están reclutando o llevando a cabo ensayos de Fase I/II en pacientes con DMD tras hacer pruebas en controles sanos (Tabla 2). Como resultado, hay más datos disponibles sobre la seguridad y tolerabilidad para NS-089/NCNP en pacientes con DMD, pero los resultados de los estudios de extensión y los ensayos EXPLORE44-OLE o ELEVATE-44-102 proporcionarán más datos de seguridad para comparar entre los tres tratamientos (Tabla 2). Algunos PMOs conjugados con péptidos, como AVI-5038, han causado degeneration tubular leve y toxicidad renal en monos dosificados con 9 mg/kg semanalmente por 4 semanas, pero estos efectos tóxicos parecen ser dependientes de la especie y la dosis [79]. Debido a que el estudio NS-089/NCNP-02 tuvo un número modesto de participantes y se llevó a cabo en un periodo corto, los resultados de los estudios de extensión en curso también proporcionarán perspectivas cruciales sobre la seguridad a largo plazo y el potencial clínico de terapias antisentido con doble objetivo
Tabla 2. Comparación de las estrategias entre AOC1044, ENTR-601-44, y NS-089/NCNP-02 y el progreso clínico actual.

En comparación con los índices de restauración de distrofina de vitolarsen que ya está aprobado (pre-tratamiento 0.3% a post-tratamiento 5.7%), los incrementos de distrofina de 24.47% para NS-089/NCNP-02 y alrededor de 20% para AOC1044 son prometedores [59, 66]. Si la distrofina producida es efectiva, estos niveles de expresión de distrofina pudieran conducir a beneficios clínicos significativos. La habilidad tanto de AOC1044 y ENTR-601-44 de orientarse al tejido muscular cardíaco es también prometedor, dada la morbilidad y mortalidad relacionadas con la miocardiopatía en pacientes con DMD.
En resumen, AOC1044, ENTR-601-44, y PGN-EDO44 representan una nueva generación de terapias de omisión de exón que usan anticuerpos o conjugaciones de péptidos para optimizar la captación de tejido y la eficiencia de entrega o suministro. NS-089/NCNP-02 logra una restauración de distrofina prometedora sin tales conjugaciones, confiando a su vez en un enfoque de doble objetivo novedoso que pudiera ofrecer beneficios específicos. A pesar de que NS-089/NCNP-02 actualmente lidera el desarrollo clínico con datos de seguridad y eficacia alentadores, los ensayos en curso y futuros de los candidatos serán esenciales para determinar su seguridad a largo plazo, eficacia cardiaca, e impacto en la función muscular.
- Abordando retos relacionados con las terapias de omisión de exón
Dada la necesidad de tratamientos que aborden la base molecular detrás de la DMD y la eficacia limitada de las terapias de ASO existentes, hay una necesidad por terapias novedosas para la DMD que muestren eficacia incrementada y restauración de distrofina. Las terapias de ASO aprobadas actualmente tienen cantidades bajas de restauración de distrofina debido al atrapamiento endosomal, liberación rápida del torrente sanguíneo, y poca captación del músculo [80]. Estas limitaciones actualmente se están abordando a través del estudio de conjugaciones de anticuerpos, como AOC1044, así como al desarrollar péptidos de penetradores celulares, como aquellos en DG9-PMO, PGN-EDO044, y ENTR-601-44 [59, 64, 75, 77]. Se necesita mayor investigación para mejorar las terapias basadas en ASO con entrega y mecanismo de orientación novedosos. Incluso la restauración parcial de los niveles de distrofina podría conducir a mejoras significativas durante las pruebas funcionales [13].
La omisión del exón 44 puede tratar hasta el 6-7% de pacientes con DMD, y es uno de varios exones que pueden ser objetivo para restaurar el marco de lectura del gen DMD. De entre las terapias de omisión de exón, la omisión del exón 51 tiene la aplicabilidad teórica más amplia, apuntando hasta el 13% de todos los pacientes con DMD [81]. Sin embargo, este es un grupo relativamente pequeño del total de la población de pacientes. Incluso después de la aprobación, cada terapia puede sólo beneficiar a una fracción de pacientes con DMD, requiriendo del desarrollo de una medicina por separado para cada mutación susceptible. Las terapias de ASO que pueden omitir uno o dos exones pueden tener el potencial de tratar 83% de pacientes con DMD (79% de todos los pacientes con deleciones, 73% de todos los pacientes con duplicaciones, y 91% de todos los pacientes con mutaciones pequeñas), pero ello deja a 17% de pacientes con mutaciones con regiones esenciales codificantes de proteína distrofina sin terapias de ASO [40]. Dado que las terapias de omisión de exón con ASO necesitan ser personalizadas para mutaciones específicas, cada nueva terapia con ASO necesita invertir tiempo y fondos en los procesos regulatorios antes de llegar a los pacientes [82].
Una opción que pudiera tratar a un grupo más grande de pacientes con DMD sería administrar múltiples terapias de omisión de exón con ASO juntas, como un ‘cóctel’ [83]. Este enfoque, aplicado a los exones 45-55, sería teóricamente aplicable hasta el 63-65% de pacientes con DMD con mutaciones de deleción, como la combinación de múltiples ASOs inducirían la omisión de múltiples exones [37, 84]. Los estudios preclínicos tienen eficiencias de multi-omisión de exón de hasta 155 para los exones 45 a 55 en modelos de ratón DMD humanizados tratados con una dosis de 1.67 μg/PMO [37]. Añadir terapias antisentido de doble objetivo en estas mezclas cóctel pudiera ser un área de estudio potencial en el futuro. Sin embargo, las mezclas de multi-omisión de exón requerirían un proceso de aprobación más complejo bajo los procesos regulatorios actuales que las terapias de una sola omisión de exón. Ello pudiera incurrir en la inversión de gastos y tiempos significativamente más altos que los tratamientos con ASO individuales.
Dependiendo de cuáles exones son omitidos, se producen distintas distrofinas truncadas, y hay diferencias funcionales entre las distrofinas dentro del marco truncadas [85]. Ciertas deleciones pudieran resultar en interacciones alteradas con otras proteínas e inestabilidad dentro de la proteína distrofina misma. Por ejemplo, las deleciones de los exones 45 a 46 en el marco a veces resultan en un fenotipo DMD, pero las deleciones de los exones 45 a 47 ó 45 a 48 a veces resultan en un fenotipo DMB más leve [34]. De ahí que medir la eficacia usando el porcentaje de restauración de distrofina pudiera no resultar necesariamente en mejoras funcionales reales en los pacientes. Esto se debe a la estructura de la proteína distrofina, con algunos dominios siendo más cruciales y menos omitibles que los otros para las interacciones de proteína del sarcolema [2]. También es importante notar que las distrofinas truncadas acortadas, como aquellas con el deleciones en los exones 45 a 55 y deleciones en los exones 3 a 9, están asociadas con un fenotipo más leve que las proteínas más grandes, lo cual añade complejidad [39, 86]. Algunas terapias de ASO actualmente aprobadas, como eteplirsen y golodirsen, tuvieron aprobaciones controversiales que estaban basadas en incrementos en la expresión de distrofina sin conclusiones definitivas sobre mejoras funcionales [30, 82, 87]. Los beneficios clínicos benéficos modestos fueron observados para estos tratamientos años después de la aprobación inicial de la FDA, con más ensayos clínicos (NCT0393430; NCT02500381) aún en curso [88, 89, 90]. Basados en estas aprobaciones y las diferentes distrofinas truncadas producidas, es posible que la eficacia de los actuales ASOs en la etapa clínica pudieran no ser completamente caracterizadas por años, incluso si estas terapias obtienen aprobación de la autoridad regulatoria.
Idealmente, las terapias moleculares para la DMD deberían restaurar la distrofina en los músculos cardíacos, esqueléticos, y respiratorios. Con los incrementos de distrofina limitados que se observaron en el tejido cardiaco después de la administración de NS-089/NCNP-02, se necesitan otros enfoques para apuntar al músculo cardiaco. La investigación de estos enfoques alternativos, como terapias génicas, terapias basadas en CRISPR, terapias basadas en células, y otros mecanismos, deberían ser continuados. Numerosos ensayos clínicos están en el camino a evaluar estos enfoques alternativos; sin embargo, la mayoría de las estrategias, aparte de aquellas que establecen nuevos propósitos para los tratamientos existentes aprobados para otras condiciones, continúan enfrentando retos regulatorios similares, con cuestiones de seguridad y eficacia únicas que contribuyen al índice de éxito en 4.6% de los ensayos clínicos [91, 92]. Por ejemplo, las terapias génicas que inicialmente pretendían superar la necesidad de dosis repetidas de tratamientos ASO existentes y estaban pensadas para ser aplicables en casi todos los pacientes con DMD, pero en lugar de eso condujeron a la implementación de criterios de exclusión tras varias reacciones inmunes severas contra las proteínas de micro distrofina extraña expresada [53, 93, 94]. Existe una necesidad por cubrir de métodos de restauración de distrofina alternativos y rentables. Los enfoques de tratamiento prometedor deberían construirse sobre los resultados existentes, así como predecir y optimizar su tolerabilidad en una población con elevados riesgos de complicaciones y necesidades de cuidado.
- Conclusiones
De manera general, NS-089/NCNP-02 es un candidato terapéutico prometedor de la DMD para la restauración de distrofina en el músculo esquelético que utiliza una estrategia de oligonucleótidos de doble objetivo. Este enfoque novedoso amplía significativamente el panorama en el diseño de terapias de ASO, lo cual pudiera mejorar la eficacia terapéutica y aplicabilidad a lo largo de un amplio rango de mutaciones. El uso de una secuencia de doble objetivo pudiera proporcionar un camino de tratamiento alternativo que evite los problemas de patente existentes y esquive la confianza en la conjugación de anticuerpos o péptidos. Cuando se combina con conjugados de anticuerpos o péptidos, esta estrategia pudiera mejorar aún más la eficiencia de entrega y potencia terapéutica. Se ha realizado un progreso considerable en la aplicación de terapia antisentido para la DMD, y NS-089/NCNP-02 muestra eficacia prometedora en los músculos esqueléticos que exceden las de las terapias de ASO aprobadas para la DMD. Las estrategias moleculares que son más aplicables ampliamente para todos los pacientes DMD pudieran mejorar significativamente los resultados a largo plazo y disminuir los costos de desarrollo clínicos para un grupo de pacientes más extenso.
Fondos
Esta investigación no recibió fondos externos.
Conflictos de interés
T. Y. es co-fundadora y comparte acciones de OligomicsTx. Inc.
Abreviaturas
DMD Distrofia muscular de Duchenne
ASO Oligonucleótido antisentido (ASO, por sus iniciales en inglés)
PMO Oligómeros de morfolino fosforodiamidato (PMOs, por sus iniciales en inglés)
EEV Vehículo de Escape Endosomal (EEV, por sus iniciales en inglés)
MTD Dosis máxima tolerada (MTD, por sus iniciales en inglés)
TfR1 Receptor 1 de anti-transferrina
DMB Distrofia muscular de Becker
Referencias
- 1. Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifiro, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- 2. Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- 3. Gao, Q.Q.; McNally, E.M. The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- 4. Salari, N.; Fatahi, B.; Valipour, E.; Kazeminina, M.; Fatahian, R.; Kiaei, A.; Shohaimi, S.; Mohammadi, M. Global prevalence of Duchenne and Becker muscular dystrophy: A systematic review and meta-analysis. J. Orthop. Surg. Res. 2022, 17, 96. [Google Scholar] [CrossRef]
- 5. Sussman, M. Duchenne Muscular Dystrophy. J. Am. Acad. Orthop. Surg. 2002, 10, 138–151. [Google Scholar] [CrossRef] [PubMed]
- 6. Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- 7. Shieh, P.B. Emerging Strategies in the Treatment of Duchenne Muscular Dystrophy. Neurotherapeutics 2018, 15, 840–848. [Google Scholar] [CrossRef]
- 8. Bushby, K.; Finkel, R.; Birnkrant, D.J.; Case, L.E.; Clemens, P.R.; Cripe, L.; Kaul, A.; Kinnett, K.; McDonald, C.; Pandya, S.; et al. Review Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010, 9, 77–93. [Google Scholar] [CrossRef]
- 9. Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; Kock, S.D.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- 10. Mercuri, E.; Bönnemann, C.G.; Muntoni, F. Muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- 11. Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- 12. Gatto, F.; Benemei, S.; Piluso, G.; Bello, L. The complex landscape of DMD mutations: Moving towards personalized medicine. Front. Genet. 2024, 15, 1360224. [Google Scholar] [CrossRef]
- 13. Godfrey, C.; Muses, S.; McClorey, G.; Wells, K.E.; Coursindel, T.; Terry, R.L.; Betts, C.; Hammond, S.; O’Donovan, L.; Hildyard, J.C.W.; et al. How much dystrophin is enough: The physiological consequences of different levels of dystrophin in the mdx mouse. Hum. Mol. Genet. 2015, 24, 4225–4237. [Google Scholar] [CrossRef] [PubMed]
- 14. Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Alman, B.A.; Apkon, S.D.; Blackwell, A.; Case, L.E.; Cripe, L.; Hadjiyannakis, S.; Olson, A.K.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef] [PubMed]
- 15. Thomas, S.; Conway, K.M.; Fapo, O.; Holtzer, C.; Meany, F.J.; Andrews, J. Time to diagnosis of Duchenne muscular dystrophy remains unchanged: Findings from the Muscular Dystrophy Surveillance, Tracking, and Research Network, 2000–2015. Muscle Nerve 2022, 66, 193–197. [Google Scholar] [CrossRef]
- 16. Abbs, S.; Tuffery-Giraud, S.; Bakker, E.; Ferlini, A.; Sejersen, T.; Mueller, C.R. Best Practice Guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul. Disord. 2010, 20, 422–427. [Google Scholar] [CrossRef] [PubMed]
- 17. Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.; Duval, B.; Bouffard, G. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef]
- 18. Emery, A.E. Clinical and molecular studies in Duchenne muscular dystrophy. Prog. Clin. Biol. Res. 1989, 306, 15–28. [Google Scholar]
- 19. Andrews, J.G.; Wahl, R.A. Duchenne and Becker muscular dystrophy in adolescents: Current perspectives. Adolesc. Health Med. Ther. 2018, 9, 53–63. [Google Scholar] [CrossRef]
- 20. Zambon, A.A.; Ayyar Gupta, V.; Ridout, D.; Manzur, A.Y.; Baranello, G.; Trucco, F.; Muntoni, F.; Tirupath, S.; Douglas, M.; McFetridge, J.; et al. Peak functional ability and age at loss of ambulation in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2022, 64, 979–988. [Google Scholar] [CrossRef]
- 21. Garg, S. Management of scoliosis in patients with Duchenne muscular dystrophy and spinal muscular atrophy: A literature review. J. Pediatr. Rehabil. Med. 2016, 9, 23–29. [Google Scholar] [CrossRef]
- 22. Hendriksen, J.G.M.; Vles, J.S.H. Neuropsychiatric disorders in males with duchenne muscular dystrophy: Frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive–compulsive disorder. J. Child Neurol. 2008, 23, 477–481. [Google Scholar] [CrossRef] [PubMed]
- 23. Banihani, R.; Smile, S.; Yoon, G.; Dupuis, A.; Mosleh, M.; Snider, A.; McAdam, L. Cognitive and Neurobehavioral Profile in Boys With Duchenne Muscular Dystrophy. J. Child Neurol. 2015, 30, 1472–1482. [Google Scholar] [CrossRef]
- 24. Houwen-van Opstal, S.L.S.; Heutinck, L.; Jansen, M.; Krom, Y.D.; Cup, E.H.C.; Hendriksen, J.G.M.; Willemsen, M.A.A.P.; Verschuuren, J.J.G.M.; Niks, E.H.; de Groot, I.J.M. Occurrence of symptoms in different stages of Duchenne muscular dystrophy and their impact on social participation. Muscle Nerve 2021, 64, 701–709. [Google Scholar] [CrossRef] [PubMed]
- 25. Mann, J.R.; Zhang, Y.; McDermott, S.; Wang, Y.; Cai, B.; Conway, K.M.; Paramsothy, P.; Royer, J.; Venkatesh, S.; Howard, J.F., Jr.; et al. Racial and Ethnic Differences in Timing of Diagnosis and Clinical Services Received in Duchenne Muscular Dystrophy. Neuroepidemiology 2023, 57, 90–99. [Google Scholar] [CrossRef]
- 26. Posner, N.; Manjelievskaia, J.; Talaga, A.K.; Richards, M.; Lew, C.R.; Merla, V.; Jimenez Alvir, J.M.; Nelson, S.F. Real-world treatment and health care utilization among patients with Duchenne muscular dystrophy by race and ethnicity in a Medicaid population. J. Manag. Care Spec. Pharm. 2025, 31, 205–213. [Google Scholar] [CrossRef] [PubMed]
- 27. Sheikh, O.; Yokota, T. Developing DMD therapeutics: A review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies. Expert Opin. Investig. Drugs 2021, 30, 167–176. [Google Scholar] [CrossRef]
- 28. Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- 29. Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- 30. Anwar, S.; Yokota, T. Golodirsen for Duchenne muscular dystrophy. Drugs Today 2020, 56, 491. [Google Scholar] [CrossRef]
- 31. Dhillon, S. Viltolarsen: First Approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- 32. Clemens, P.R.; Previtera, M.L.; Crozier, R.A.; Magnus, L.; Hoffman, E.; Komaki, H.; Aoki, Y.; Rao, V.K. Brogidirsen, an Investigational exon 44 Skipping Agent for the Treatment of Duchenne Muscular Dystrophy: Clinical Trial Design (Phase 2); Muscular Dystrophy Association National Office: Chicago, IL, USA, 2024. [Google Scholar]
- 33. Adkin, C.F.; Meloni, P.L.; Fletcher, S.; Adams, A.M.; Muntoni, F.; Wong, B.; Wilton, S.D. Multiple exon skipping strategies to by-pass dystrophin mutations. Neuromuscul. Disord. 2012, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- 34. Findlay, A.R.; Wein, N.; Kaminoh, Y.; Taylor, L.E.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. Clinical phenotypes as predictors of the outcome of skipping around DMD exon 45. Ann. Neurol. 2015, 77, 668–674. [Google Scholar] [CrossRef]
- 35. Anwar, S.; Yokota, T. The Dysferlinopathies Conundrum: Clinical Spectra, Disease Mechanism and Genetic Approaches for Treatments. Biomolecules 2024, 14, 256. [Google Scholar] [CrossRef] [PubMed]
- 36. Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-conjugate antisense based splice-correction for Duchenne muscular dystrophy and other neuromuscular diseases. EBioMedicine 2019, 45, 630–645. [Google Scholar] [CrossRef]
- 37. Echigoya, Y.; Lim, K.R.Q.; Melo, D.; Bao, B.; Trieu, N.; Mizobe, Y.; Maruyama, R.; Mamchaoui, K.; Tanihata, J.; Aoki, Y.; et al. Exons 45–55 Skipping Using Mutation-Tailored Cocktails of Antisense Morpholinos in the DMD Gene. Mol. Ther. 2019, 27, 2005–2017. [Google Scholar] [CrossRef]
- 38. Min, Y.-L.; Chemello, F.; Li, H.; Rodriguez-Caycedo, C.; Sánchez-Ortiz, E.; Mireault, A.A.; McAnally, J.R.; Shelton, J.M.; Zhang, Y.; Bassel-Duby, R.; et al. Correction of Three Prominent Mutations in Mouse and Human Models of Duchenne Muscular Dystrophy by Single-Cut Genome Editing. Mol. Ther. 2020, 28, 2044–2055. [Google Scholar] [CrossRef]
- 39. Nakamura, A.; Fueki, N.; Shiba, N.; Motoki, H.; Miyazaki, D.; Nishizawa, H.; Echigoya, Y.; Yokota, T.; Aoki, Y.; Takeda, S.I. Deletion of exons 3−9 encompassing a mutational hot spot in the DMD gene presents an asymptomatic phenotype, indicating a target region for multiexon skipping therapy. J. Hum. Genet. 2016, 61, 663–667. [Google Scholar] [CrossRef]
- 40. Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; van Deutekom, J.C.; Heemskerk, H.; t’Hoen, P.; de Kimpe, S. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef]
- 41. Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- 42. Petersen, M.; Nielsen, C.B.; Nielsen, K.E.; Jensen, G.A.; Bondensgaard, K.; Singh, S.K.; Rajwanshi, V.K.; Koshkin, A.A.; Dahl, B.M.; Wengel, J. The conformations of locked nucleic acids (LNA). J. Mol. Recognit. 2000, 13, 44–53. [Google Scholar] [CrossRef]
- 43. Hill, A.C.; Hall, J. The MOE Modification of RNA: Origins and Widescale Impact on the Oligonucleotide Therapeutics Field. Helv. Chim. Acta 2023, 106, e202200169. [Google Scholar] [CrossRef]
- 44. Sabrina Haque, U.; Kohut, M.; Yokota, T. Comprehensive review of adverse reactions and toxicology in ASO-based therapies for Duchenne Muscular Dystrophy: From FDA-approved drugs to peptide-conjugated ASO. Curr. Res. Toxicol. 2024, 7, 100182. [Google Scholar] [CrossRef]
- 45. Devi, G.R. Delivery of Phosphorodiamidate Morpholino Antisense Oligomers in Cancer Cells. Methods Mol. Biol. 2009, 542, 351–361. [Google Scholar] [PubMed]
- 46. Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- 47. Maksudov, F.; Kliuchnikov, E.; Pierson, D.; Ujwal, M.L.; Marx, K.A.; Chanda, A.; Barsegov, V. Therapeutic phosphorodiamidate morpholino oligonucleotides: Physical properties, solution structures, and folding thermodynamics. Mol. Ther. Nucleic Acids 2023, 31, 631–647. [Google Scholar] [CrossRef] [PubMed]
- 48. Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.B.; Weller, D.D. Resistance of Morpholino Phosphorodiamidate Oligomers to Enzymatic Degradation. Antisense Nucleic Acid Drug Dev. 1996, 6, 267–272. [Google Scholar] [CrossRef]
- 49. Moulton, J.D.; Jiang, S. Gene Knockdowns in Adult Animals: PPMOs and Vivo-Morpholinos. Molecules 2009, 14, 1304–1323. [Google Scholar] [CrossRef]
- 50. Palacio-Castañeda, V.; Brock, R.; Verdurmen, W.P.R. Generation of Protein-Phosphorodiamidate Morpholino Oligomer Conjugates for Efficient Cellular Delivery via Anthrax Protective Antigen. Methods Mol. Biol. 2022, 2434, 129–141. [Google Scholar]
- 51. Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- 52. Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- 53. ang, A.; Yokota, T. Is Duchenne gene therapy a suitable treatment despite its immunogenic class effect? Expert Opin. Drug Saf. 2025, 24, 395–411. [Google Scholar] [CrossRef] [PubMed]
- 54. Ersöz, E.; Demir-Dora, D. Unveiling the potential of antisense oligonucleotides: Mechanisms, therapies, and safety insights. Drug Dev. Res. 2024, 85, e22187. [Google Scholar] [CrossRef] [PubMed]
- 55. Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- 56. Stahl, M.; Zhu, Y.; Goel, V.; Leung, L.; Tami, Y.; Hardin, T.; Etxaniz, U.; Kovach, P.; Herzog, J.; Hughes, S.; et al. AOC 1044 as a Novel Therapeutic Approach for DMD Patients Amenable to Exon 44 Skipping: EXPLORE44TM Phase 1/2 Healthy Volunteer Data; Muscular Dystrophy Association National Office: Orlando, FL, USA, 2024. Available online: https://clinicaltrials.gov/ct2/show/NCT05670730 (accessed on 16 June 2025).
- 57. Cochran, M.; Marks, I.; Albin, T.; Aras, D.; Kovach, P.; Darimont, B.; Huang, H.; Etxaniz, U.; Kwon, H.W.; Shi, Y.; et al. Structure–Activity Relationship of Antibody–Oligonucleotide Conjugates: Evaluating Bioconjugation Strategies for Antibody–Phosphorodiamidate Morpholino Oligomer Conjugates for Drug Development. J. Med. Chem. 2024, 67, 14868–14884. [Google Scholar] [CrossRef]
- 58. Wilton-Clark, H.; Yokota, T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics 2023, 15, 778. [Google Scholar] [CrossRef]
- 59. Etxaniz, U.; Marks, I.; Albin, T.; Diaz, M.; Bhardwaj, R.; Anderson, A.; Tyaglo, O.; Hoang, T.; Missinato, M.A.; Svensson, K.; et al. AOC 1044 induces exon 44 skipping and restores dystrophin protein in preclinical models of Duchenne muscular dystrophy. Nucleic Acids Res. 2025, 53, gkaf241. [Google Scholar] [CrossRef]
- 60. Nguyen, Q.; Yokota, T. Antisense oligonucleotides for the treatment of cardiomyopathy in Duchenne muscular dystrophy. Am. J. Transl. Res. 2019, 11, 1202–1218. [Google Scholar]
- 61. Entrada Therapeutics. Entrada Therapeutics Announces FDA Removal of Clinical Hold on ENTR-601-44. 2025. Available online: www.entradatx.com (accessed on 16 June 2025).
- 62. Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Cross, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- 63. Sahni, A.; Qian, Z.; Pei, D. Cell-Penetrating Peptides Escape the Endosome by Inducing Vesicle Budding and Collapse. ACS Chem. Biol. 2020, 15, 2485–2492. [Google Scholar] [CrossRef]
- 64. Oldham, M.; Estrella, N.; Kumar, A.; Hicks, A.; Brennan, C.; Blake, S.; Li, X.; Pathak, A.; Kheirabadi, M.; Dougherty, P.; et al. 433P Therapeutic potential of ENTR-601-44, an Endosomal Escape Vehicle (EEVTM)—Oligonucleotide Conjugate for the treatment of exon 44 skip amenable DMD. Neuromuscul. Disord. 2024, 43, 104441.304. [Google Scholar] [CrossRef]
- 65. Wahab, E. A Study to Investigate How ENTR-601-44 Behaves in the Body, the Safety and How Well Tolerated Different Increasing Amounts of the Drug ENTR-601-44 Are When Given to Healthy Male Volunteers; ISRCTN: London, UK, 2024; MAC Clinical Research; Available online: http://isrctn.com/ (accessed on 16 June 2025).
- 66. Komaki, H.; Takeshita, E.; Kunitake, K.; Ishizuka, T.; Shimizu-Motohashi, Y.; Ishiyama, A.; Sasaki, M.; Yonee, C.; Maruama, S.; Hida, E.; et al. Phase 1/2 trial of brogidirsen: Dual-targeting antisense oligonucleotides for exon 44 skipping in Duchenne muscular dystrophy. Cell Rep. Med. 2025, 6, 101901. [Google Scholar] [CrossRef] [PubMed]
- 67. Nippon Shinyaku Co., Ltd. FDA Grants Breakthrough Therapy Designation to NS-089/NCNP-02 for the Treatment of Duchenne Muscular Dystrophy. Nippon Shinyaku Co., Ltd. 28 July 2023. Available online: https://www.ncnp.go.jp/topics/2022/20220317e.html (accessed on 16 June 2025).
- 68. Komaki, H.; Takeshita, E.; Kunitake, K.; Shimizu-Motohashi, Y.; Sasaki, M.; Yonee, C.; Maruyama, S.; Hida, E.; Matsubara, D.; Hatakeyama, T.; et al. P.123 A Phase I/II study of NS-089/NCNP-02, Exon 44 skipping drug, in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2022, 32, S99–S100. [Google Scholar] [CrossRef]
- 69. Shimo, T.; Maruyama, R.; Yokota, T. Designing Effective Antisense Oligonucleotides for Exon Skipping. In Duchenne Muscular Dystrophy: Methods and Protocols; Bernardini, C., Ed.; Humana Press: New York, NY, USA, 2018; Available online: http://www.springer.com/series/7651 (accessed on 16 June 2025).
- 70. Watanabe, N.; Tone, Y.; Nagata, T.; Masuda, S.; Saito, T.; Motohashi, N.; Takagaki, K.; Aoki, Y.; Takeda, S. Exon 44 skipping in Duchenne muscular dystrophy: NS-089/NCNP-02, a dual-targeting antisense oligonucleotide. Mol. Ther. Nucleic Acids 2023, 34, 102034. [Google Scholar] [CrossRef]
- 71. Komaki, H.; Takeshima, Y.; Matsumura, T.; Ozasa, S.; Funato, M.; Takeshita, E.; Iwata, Y.; Yajima, H.; Egawa, Y.; Toramoto, T.; et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol. 2020, 7, 2393–2408. [Google Scholar] [CrossRef]
- 72. Ishizuka, T.; Komaki, H.; Asahina, Y.; Nakamura, H.; Motohashi, N.; Takeshita, E.; Shimizu-Motohashi, Y.; Ishiyama, A.; Tonee, C.; Maruyama, S.; et al. Systemic administration of the antisense oligonucleotide NS<-089/NCNP-02 for skipping of exon 44 in patients with Duchenne muscular dystrophy: Study protocol for a phase I/II clinical trial. Neuropsychopharmacol. Rep. 2023, 43, 277–286. [Google Scholar]
- 73. Broomfield, J.; Abrams, K.; Latimer, N.; Guglieri, M.; Rutherford, M.; Crowther, M. Natural history of Duchenne muscular dystrophy in the United Kingdom: A descriptive study using the Clinical Practice Research Datalink. Brain Behav. 2023, 13, e3331. [Google Scholar] [CrossRef]
- 74. Tulangekar, A.; Sztal, T.E. Inflammation in Duchenne Muscular Dystrophy–Exploring the Role of Neutrophils in Muscle Damage and Regeneration. Biomedicines 2021, 9, 1366. [Google Scholar] [CrossRef]
- 75. Holland, A.; Lonkar, P.; Sweeney, C.; Zhang, H.; Svenstrup, N.; Gibbons, C.; Xu, L.; Joy, J.; Goyal, J. P27 Three novel enhanced delivery Oligonucleotide candidates for Duchenne muscular dystrophy mediate high levels of exon 53, 45, and 44 skipping. Neuromuscul. Disord. 2023, 33, S103–S104. [Google Scholar] [CrossRef]
- 76. Mellion, M.; Larkindale, J.; Lonkar, P.; Goyal, J.; Holland, A.; Foy, J.; Garg, B.; Yu, S.; Frank, A.; Abbott, C.; et al. PGN-EDO51, an Enhanced Delivery Oligonucleotide (EDO) for the Treatment of Duchenne Muscular Dystrophy (DMD): Results of a Phase 1 Study in Healthy Volunteers. American Academy of Neurology 2023 Annual Meeting. American Academy of Neurology. 2023. Available online: https://www.aan.com/MSA/Public/Events/AbstractDetails/54382 (accessed on 25 June 2025).
- 77. Shah, M.N.A.; Wilton-Clark, H.; Haque, F.; Powell, B.; Sutanto, L.E.; Maradiya, R.; Zhabyeyev, P.; Roshimi, R.R.; Anwar, S.; Aslesh, T.; et al. DG9 boosts PMO nuclear uptake and exon skipping to restore dystrophic muscle and cardiac function. Nat. Commun. 2025, 16, 4477. [Google Scholar] [CrossRef]
- 78. Shang, M.; Wu, Y.; Wang, Y.; Cai, Y.; Jin, J.; Yang, Z. Dual antisense oligonucleotide targeting miR-21/miR-155 synergize photodynamic therapy to treat triple-negative breast cancer and inhibit metastasis. Biomed. Pharmacother. 2022, 146, 112564. [Google Scholar] [CrossRef]
- 79. Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. et Biophys. Acta (BBA) Biomembr. 2010, 1798, 2296–2303. [Google Scholar] [CrossRef]
- 80. Haque, U.S.; Yokota, T. Enhancing Antisense Oligonucleotide-Based Therapeutic Delivery with DG9, a Versatile Cell-Penetrating Peptide. Cells 2023, 12, 2395. [Google Scholar] [CrossRef]
- 81. Waldrop, M.A.; Yaou, R.B.; Lucas, K.K.; Martin, A.S.; O’Rourke, E.; Filnemus; Ferlini, A.; Muntoni, F.; Leturcq, F.; Tuffery-Giraud, S.; et al. Clinical Phenotypes of DMD Exon 51 Skip Equivalent Deletions: A Systematic Review. J. Neuromuscul. Dis. 2020, 7, 217–229. [Google Scholar] [CrossRef] [PubMed]
- 82. Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- 83. Lim, K.R.Q.; Woo, S.; Melo, D.; Huang, Y.; Dzierlega, K.; Shah, M.N.A.; Aslesh, T.; Roshmi, R.R.; Echigoya, Y.; Maruyama, R.; et al. Development of DG9 peptide-conjugated single- and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2112546119. [Google Scholar] [CrossRef] [PubMed]
- 84. Béroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizaed, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef] [PubMed]
- 85. Van Deutekom, J.C.T.; Van Ommen, G.J.B. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef]
- 86. Anthony, K.; Cirak, S.; Torelli, S.; Tasca, G.; Feng, L.; Arechavala-Gomeza, V.; Armaroli, A.; Guglieri, M.; Straathof, C.S.; Verschuuren, J.J.; et al. Dystrophin quantification and clinical correlations in Becker muscular dystrophy: Implications for clinical trials. Brain 2011, 134, 3547–3559. [Google Scholar] [CrossRef]
- 87. Heo, Y.-A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- 88. Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenyalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- 89. McDonald, C.M.; Shieh, P.B.; Abdel-Hamid, H.Z.; Connolly, A.M.; Ciafaloni, E.; Wagner, K.R.; Goemans, N.; Mercuri, E.; Khan, N.; Koenig, E.; et al. Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial. J. Neuromuscul. Dis. 2021, 8, 989–1001. [Google Scholar] [CrossRef]
- 90. Servais, L.; Mercuri, E.; Straub, V.; Guglieri, M.; Seferian, A.M.; Scoto, M.; Leone, D.; Koenig, E.; Khan, N.; Dugar, A.; et al. Long-Term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-in-human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial. Nucleic Acid Ther. 2022, 32, 29–39. [Google Scholar] [CrossRef] [PubMed]
- 91. Tang, A.; Yokota, T. Duchenne muscular dystrophy: Promising early-stage clinical trials to watch. Expert Opin. Investig. Drugs 2024, 33, 201–217. [Google Scholar] [CrossRef] [PubMed]
- 92. Dovari, A.; Inuganti, B.; Nadimpally, J.; Vatturi, S.M.; Hyderboini, R.; Goyal, R. PRO38 Twenty Years of Clinical Trials in Duchenne Muscular Dystrophy: A Low Clinical Drug Development Success. Value Health 2021, 24, S204. [Google Scholar] [CrossRef]
- 93. Lek, A.; Wong, B.; Keeler, A.; Blackwood, M.; Ma, K.; Huang, S.; Sylvia, K.; Batista, R.; Artinian, R.; Kokoski, D.; et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 389, 1203–1210. [Google Scholar] [CrossRef]
- 94. Bönnemann, C.G.; Belluscio, B.A.; Braun, S.; Morris, C.; Singh, T.; Muntoni, F. Dystrophin Immunity after Gene Therapy for Duchenne’s Muscular Dystrophy. N. Engl. J. Med. 2023, 388, 2294–2296. [Google Scholar] [CrossRef]
Descargo de responsabilidad / Nota del editor: Las declaraciones, opiniones, y datos contenidos en todas las publicaciones son únicamente aquellas del autor (es) y contribuyente(s) individual(es) y no de MDPI y/o el editor(es). MDPI y/o editor(es) se deslindan de responsabilidad por cualquier daño a personas o propiedad resultante de las ideas, métodos, instrucciones, o productos referidos en el contenido.
© 2025 por los autores. Licencia de MDPI, Basel. Suiza. Este artículo es un artículo de acceso abierto distribuido bajo los términos y condiciones de la licencia Creative Commons Attribution (CC BY) (https://creativecommons.org/licenses/by/4.0/).
Esta publicación está escrita en español para promover el acceso de la comunidad a información y actualizaciones. La información que se comparte tiene como objetivo el interés y la concientización general y, por lo tanto, no debe ser considerada ni interpretada como consejo médico o legal. Se trata de información publicada abiertamente. La Fundación Akari no promociona ninguna empresa, producto o tratamiento específico. Le recomendamos que consulte la fuente original y tome decisiones informadas.
Para leer el artículo original en inglés consulte:
Si desea saber más información acerca de este artículo de investigación, contáctese directamente con la fuente original del mismo.